Статья:




«ГЕНЕТИЧЕСКИЕ АСПЕКТЫ БОЛЕЗНИ ВИЛЬСОНА-КОНОВАЛОВА»
Секция: 2. Биологические науки
Выходные данные
Курочкина И.М. «ГЕНЕТИЧЕСКИЕ АСПЕКТЫ БОЛЕЗНИ ВИЛЬСОНА-КОНОВАЛОВА» // Молодежный научный форум: Естественные и медицинские науки: электр. сб. ст. по мат. XXIII междунар. студ. науч.-практ. конф. № 4(22). URL: https://nauchforum.ru/archive/MNF_nature/4(22).pdf (дата обращения: 29.07.2026)
Лауреаты определены. Конференция завершена
Эта статья набрала 2829 голосов
Мне нравится2829
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников

XXIII Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки»
«ГЕНЕТИЧЕСКИЕ АСПЕКТЫ БОЛЕЗНИ ВИЛЬСОНА-КОНОВАЛОВА»
Курочкина Ирина Михайловна
студент 1 курса, 72 группы лечебного факультета, Первого МГМУ им. И.М. Сеченова, РФ, г. Москва
Валова Татьяна Ивановна
научный руководитель, канд. биол. наук, доц. кафедры биологии и общей генетики Первого МГМУ им. И.М. Сеченова, РФ, г. Москва
Дегтяревская Татьяна Юрьевна
Изучив автореферат диссертации на соискание учебной степени кандидата медицинских наук Г.Я. Левиной под названием «Морфологические изменения в печени при гепато-церебральной дистрофии (болезни Вильсона-Коновалова)», 1968 года издания дается следующее определение болезни Вильсона. Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу (A. André, L. Van Bogaert, 1950; A. Bearn, 1957, 1960, 1961; A. Sass-Kortsak, 1959) генетически обусловленных патологических признаков, которые появляются нарушениями обмена меди (J. Cummings, 1948, 1959, J. Scheinberg, D. Gitlin, 1952, Н.В. Коновалов, 1960 и др.) и белков (L. Uzman, D. Denny-Brown, В.П. Петрунова, 1960, Л.М. Алиева, 1963, А.А. Миттельштедт, Л.К. Бауман, В.П. Бархатова, 1965 и др.).
Болезни Вильсона-Коновалова в настоящее время дают следующее определение. Гепатолентикулярная дегенерация или болезнь Вильсона — аутосомно-рецессивное полисистемное заболевание, обусловленное избыточным накоплением меди в организме и токсичным действием меди, характеризующееся сочетанным поражением паренхиматозных органов, прежде всего печени и головного мозга, преимущественно подкорковых ядер. (Centre Hépato-Biliaire — Hôpital Universitaire Paul Brousse, la maladie de Wilson, France, 08.10.2014 года, Canadian liver foundation, Wilson disease, Canada, 2014 года; Федеральные клинические рекомендации по диагностике и лечению болезни Вильсона-Коновалова (гепатолентикулярная дегенерация), Москва, 2013 год).

Рисунок 1. Аутосомно-рецессивное наследование заболеваний
Согласно Brewer G.J. 1999, частота гепатолентикулярной дегенерации составляет 1—3 человека на 100 тысяч населения с неравномерным географическим и этническим распространением. Взяв данные 1999 года, я заметила прогрессирование этой болезни среди населения.
По данным Centre Hépato-Biliaire — Hôpital Universitaire Paul Brousse, la maladie de Wilson, France, 08.10.2014 года, Canadian liver foundation, Wilson disease, Canada, 2014 года, это заболевание поражает одного из 30 000 человек. Пронаблюдав за прогрессивностью заболевания, мне стало интересно, насколько в настоящее время опасна эта болезнь и, выяснить причины её возникновения.
Впервые о болезни Вильсона-Коновалова стало известно в 1883 году немецкими врачами C. Westphal и A. Strumpell. Затем подробнее были изучены проявления этого заболевания и способы лечения, а позже, в 1985 году, была выявлена мутация (дефект) в гене (ATP7B), который отвечает за развитие гепатолентикулярной дегенерации. Этот ген расположен на 13 хромосоме и кодирует белок (транспортирующий медь АТФ-азный протеин Р-типа), участвующий в организме в процессе транспорта ионов меди.
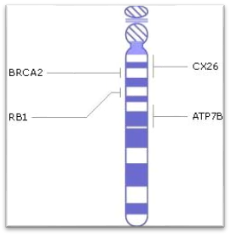
Рисунок 2. Ген ATP7B локализованный на длинном плече 13ой хромосоме
Болезнь Вильсона носит своё название в честь английского невролога Сэмюеля Вильсона, опубликовавшего свою работу в 1912 году, в которой дал описание клинической и патологоанатомической картины нового заболевания. Оно характеризовалось сочетанным поражением печени и мозга, а так же заболевание проявлялось сначала в детстве и в дальнейшем прогрессировало. Морфологически определялись двусторонние изменения чечевицеобразных ядер, а в печени во всех случаях — цирроз. В том же году Hall объединил прогрессирующую лентикулярную дегенерацию Вильсона и псевдосклероз Вестфаля-Штрюмпеля в заболевание — гепатолентикулярную дегенерацию.
В России болезнью Вильсона-Коновалова занимался в первую очередь один из крупнейших отечественных неврологов Николай Васильевич Коновалов и его школа. Тщательное изучение клиники и морфологии гепатолентикулярной дегенерации дали возможность Николаю Васильевичу создать подробную оригинальную классификацию болезни (1948, 1960 года), а также показать, что патологические изменения в мозге не ограничиваются чечевицеобразными ядрами, а носят диффузный характер. Из-за этого Н.В. Коновалов считал, что более точно характеризует процесс новое название болезни — гепатоцеребральная дистрофия.
Болезнь Вильсона-Коновалова вызывается мутацией, которая в свою очередь вызывает отсутствие или нехватки белка ATP7B, расположенного на длинном плече 13 хромосомы (13q14.3). Чаще всего эта мутация поражает клетки печени, мозга, почек. Из-за недостатка этого белка нарушается работа медь-транспортирующей АТФ-азы.
Рисунок 3. Аномальный белок, кодирующийся геном ATP7B
Она осуществляет внутриклеточный транспорт меди в гепатоцитах (клетках печени) и участвует в выведении с желчью излишков меди, а также включает её в молекулу церулоплазмина (Горбункова В.Н. и другие, 2002 год; Иллариошкин С.Н. и другие, 2002 год; Thomas G.R. и другие, 1995 год).
Рисунок 4. Структура белка церулоплазмина
Это приводит к гипоцерулоплазминемии (биохимический маркер заболевания), а именно к увеличению концентрации в крови не связной меди с церулоплазмином, а также к развитию хронической медной интоксикации, накоплению меди в различных тканях и органах, прежде всего, как уже выше упоминалось, в печени, мозге, почках, роговице (кольца Кайзера-Флейшера).
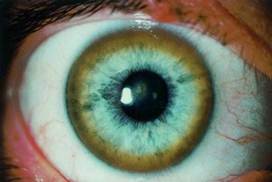
Рисунок 5. Кольцо Кайзера-Флейшера
Чтобы болезнь начала проявляться, человек должен обязательно иметь не менее двух дефектных генов, по одному от каждого родителя, иначе болезнь не будет проявляться.
Ген АТР7B состоит из чередования между собой 21 экзона и интрона. Этот ген проявляется в клетках печени (гепатоцитах), мозга, почках, лимфоузлах. Точковые мутации гена АТР7В являются характерной чертой для болезни Вильсона-Коновалова, а так же мелкие инсерции и делеции, которые вызывают сдвиг рамки считывания. В наше время известно свыше 280 различных мутаций этого белка. Из них больше всего распространена миссенс-мутация His1069Gln (Карунас А.С., 1999 года, Иллариошкин С.Н. И другие, 2002 год), которая наблюдается в европейских популяциях и России. Гепатолентикулярная дегенерация характеризуется полиморфизмом в отношении как неврологических, так и соматических проявлений. До сих пор нет объяснений множеству разных проявлений и тяжести течения при данном генетическом заболевании и кроме того выраженному внутрисемейному полиморфизму.
При выполнении своей работы я обратила внимание на молекулярно-генетическое исследование болезни Вильсона-Коновалова в республике Башкортостан. Выявлено 8 различных мутаций в гене АТР7В на сегодняшний момент. Впервые обнаружены две не описанные ранее мутации: Ala718Pro и Lys1315_Arg1316delinsGlu. С высокой частотой (10.7 %) была выявлена мутация Lys1315_Arg1316delinsGlu. Опираясь на автореферат диссертации на соискание ученой степени кандидата медицинских наук «Гено-фенотипические корреляции при болезни Вильсона в Республике Башкортостан», Магжанова А.Р., Уфа, 31.05.2007 год, впервые в России были обнаружены ранее открытые мутации Leu1305Pro и Met769fs. Впервые придали особое значение влиянию различных мутаций в гене АТР7В на клинические проявления гепатолентикулярной дегенерации, так, гомозиготы с мутацией Hisl069Gln характеризуются более мягким течением заболевания с поздним её проявлением, у пациентов с мутацией Lys1315_Arg1316delinsGlu отмечено тяжелое течение болезни, ранняя манифестация и выраженные нарушения функций печени. Установление мутаций на 83,93 % хромосом повышает эффективность молекулярно-генетического метода диагностики заболевания.
Выявление следующей по частоте мутации Lys1315_Arg1316delinsGlu в гене АТР7В дает рациональное объяснение вновь появляющихся пациентов с болезнью Вильсона-Коновалова после проведения поиска самой частой мутации His1069Gln в 14ом экзоне. Выявленные данные с мутантными аллелями в гене АТР7В повышают лечебно-диагностической и медикогенетической помощи. Мутации в гене АТР7В ассоциированы с определенными гаплотипами полиморфных ДНК-локусов D13S316, D13S133, D13S228.
Чтобы провести анализ полиморфных ДНК-локусов D13S316, D13S133 и D13S228 использовали метод полимеразной цепной реакции (ПЦР). Этот метод основывает на многочисленном выборочном копировании определенного участка дезоксирибонуклеиновой кислоты (ДНК) с помощью ферментов. ПЦР проводят в искусственных условиях. Данный метод осуществляется, если скопированный выбранный участок присутствует в исследованном образце. Для проведения метода полимеразной цепной реакции использовали выделенную методом фенольно-хлороформной экстракции по Mathew С.С. (1984) из 8 мл крови ДНК из лейкоцитов периферической крови.
Поиск на наличие мутаций и полиморфизмов в девятнадцати экзонах (2—20) гена АТР7В в образцах ДНК проводили при помощи метода анализа конформационного полиморфизма однонитевой ДНК (SSCP) (Onta M. et. al., 1989 год). Данный метол проводился на автоматическом секвенаторе ABI Prism модель 310 (Applied Biosystems). Чтобы подтвердить результаты секвенирования использовали метод полиморфизма длин рестрикционных фрагментов (ПДРФ). Данный метод исследования геномной ДНК основан на разрезании ДНК с помощью эндонуклеаз рестрикции, а после этого производится анализ размеров образующихся рестриктов (фрагментов) путем ДНК электрофореза. Во время проведения SSCP-анализа экзонов и рядом располагающихся интронных областей гена АТР7В были найдены изменения электрофоретической подвижности однонитевой ДНК в 8 экзонах (3, 6, 13, 14, 15, 16, 18, 19).
В четырнадцатом экзоне было обнаружено 4 вида изменений. При дальнейшем секвенировании были определены две мутации: His1069Gln и Glu1064Lys. Так, например, трансверсия цитозина (C) на аденин (A) в положении 3207 приводит к замене гистидина (His) на глутамин (Glu) в положении 1069 белка АТР7В. Данная мутация была установлена в гомозиготном состоянии у 5 бальных с гепатолентикулярной дегенерацией из четырех семей, а в гетерозиготном состоянии — у 25 пациентов из 19 семей в Республике Башкортостан.
В мутации Glu1064Lys происходит транзиция гуанина (G) на аденин (А) в положении 3190, что приводит к замене глутаминовой кислоты (Glu) на лизин (Lys) в положении 1064 белка ATP7B. Данная мутация определена секвенированием, также была подтверждена ПДРФанализом с использованием рестриктазы MnlI в стандартных условиях. Мутацию Glu1064Lys обнаружили у 5 пациентов из 4 семей в гетерозиготном состоянии, а именно у трех человек русского и одного человека русско-литовского происхождений. Отмечу, что в двух семьях данная мутация найдена в компаунд-гетерозиготном состоянии в сочетании с мутацией His1069Gln на 2ой хромосоме. До этого мутация Glu1064Lys была обнаружена с небольшой частотой у турков (Figus A. et. al., 1995 год), венгров (Folhoffer F. et. al., 2003 год) и чехов (Vrabelova S. et. al., 2005 год).
При скрининге девятнадцатого экзона гена АТР7В при помощи SSCP-анализа у пациентов с гепатолентикулярной дегенерацией обнаружено три типа изменений, связанные с подвижностью однонитевой ДНК. При секвенировании девятнадцатого экзона гена АТР7В у образцов с первым типом изменения подвижности однонитевой ДНК была обнаружена замена тимина (T) на цитозин (C) в положении 3914 (3914Т→С). Данная мутация приводит к замене аминокислоты лейцин (Leu) на пролин (Pro) в положении 1305 (Leu1305Pro). Мутация Leu1305Pro подтвердили при помощи ПДРФ-анализа с использованием рестриктазы MspI в стандартных условиях. Эта мутация найдена в 2х семьях русской и русско-немецкой этнической принадлежностей в компаунд-гетерозиготном состоянии, а также вместе с мутацией His1069Gln. Эта мутация была ранее описана у пациентов немецкого, французского и чешского происхождений (Genschel J., Czlonkowska A., 2001 год; Vrabelova S. et. al., 2005 год).
Секвенирование образцов ДНК со вторым и третьим изменениями подвижности однонитевой ДНК в девятнадцатом экзоне гена АТР7В больных гепатолентикулярной дегенерацией выявило делецию 3х нуклеотидов: цитозина (C) и аденина(A) в положениях 3942—3943, а так же гуанина — в положении 3947 в гетеро- и гомозиготном состоянии. Из-за комбинации 2х близь лежащих делений произошло нарушение синтеза аминокислот, но аминокислота серии не изменилась, потому что произошла аналогичная замена кодона ТСС на ТСА. Из-за этого вместо делетировавшихся 2х аминокислот лизина (Lys) и аргинина (Arg) в результате чего образовалась одна глутаминовая кислота (Glu). В общем делетировалось три нуклеотида и дальнейшего сдвига рамки считывания не наблюдалось.
Нормальная последовательность:
3889 СТТ ТСС AAG AGG ACT GTC CGA AGG ATA
1313 Leu; Ser; Lys; Arg; Thr; Vаl; Arg; Arg; Ile
Измененная последовательность:
3889 СТТ TCA CAG ACT GTC CGA AGG ATA
1313 Leu; Ser; Gln; Thr; Val; Arg; Arg; Ile
Данная делеция ранее в литературе нигде не описывалась. Мутация затрагивает область гена, который кодирует трансмембранный домен 7, и, скорее всего, приводит к нарушению медь-транспортирующей функции АТФ-азы. Мутация Lys1315_Arg_1316delinsGlu установлена у одного больного в гомозиготном состоянии, а также у четырех — в гетерозиготном.
Помимо приведенных выше мутаций, было обнаружено два типа изменений подвижности однонитевой ДНК при SSCP-анализе 8го экзона гена ATP7B у пациентов с болезнью Вильсона из республики Башкортостан. При дальнейшем секвенировании образца с первым типом изменения подвижности выявлена замена гуанина (G) на цитозин (С) в положении 2152, приводящая к замене аминокислоты аланин (Ala) на пролин (Pro) в положении 718 белка АТР7В. Для подтверждения мутации Аlа718Рго был проведен ПДРФ-анализ с помощью рестриктазы НaeIII в стандартных условиях. Нет литературы об описании ранее данной мутации в других популяциях. Данная область гена АТР7В кодирует участок трансмембранного домена Т2, сравнительно не далеко лежащего от домена ТЗ, и мутации в нем, возможно приводят к нарушению транспорта меди. Мутация Аlа718Рго обнаружена у пациента русской этнической принадлежности в компаунд-гетерозиготном состоянии вместе с мутацией Glu1064Lys.
При секвенировании 2го типа изменений в восьмом экзоне гена АТР7В у больных болезнью Вильсона-Коновалова выявлена в гетерозиготном состоянии инсерция цитозина (C) в положении 2304, приводящая к сдвигу рамки считывания, а именно начиная с аминокислоты метионин (Met) в положении 769 (Met769fs). У 2х сестер метисного происхождения была выявлена данная мутация в гетерозиготном состоянии. До этого мутацию Met769fs находили в разных популяциях, а именно: у русских, французов, англичан, итальянцев, чехов, поляков, северо-американцев европейского происхождения, тайванцев, японцев (Thomas G. et. al., 1995 год; Shah A. et. al., 1997 год; Tsai С. et. al., 1998 год; Shimizu N. et. al., 1999 год; Lee С. et. al., 2000 год; Loudianos G. et. al., 2003; Vrabelova S. et. al., 2005 год). Скорее всего, представленный участок нуклеотидной последовательности (СССССС) представляет собой «горячую точку» в мутационных изменениях.
С помощью найденных новых мутаций и вновь описанных ранее мутаций, в результате молекулярно-генетического исследования мутации были выявлены на 83,9 % хромосом. У 21 из 28 семей установили мутацию в гене АТР7В на обеих хромосомах, помимо этого, в шести семьях была определена мутация на одной хромосоме. По этнической принадлежности все выявленные семьи распределились следующим образом 11 русских, 19 татарских, 5 башкирских, 3 чувашских и в 11 семьях имел место межнациональный брак родителей пробандов, по данным ГМЦ Росстата на момент переписи населения 2002 года, исходя из численности народов разной национальности и количества живых больных Республики Башкортостан. В изоэтнических семьях, распространение болезни Вильсона составляет среди русских 1,186339 (0,54*10-5), среди татар 1,110078 (0,91*10-5), среди башкир 1,407101 (0,24*10-5), среди чувашей 1,29329 (3,41*10-5). В соответствии с принятой в нашей стране клинической классификацией болезни Вильсона-Коновалова, предложенной Н.В. Коноваловым в 1960 году, пациенты распределились следующим образом: дрожательно-ригидная форма (28 больных), затем дрожательная (20 больных), следом преневрологическая или висцеральная форма (12 больных) и, в конце, пресимптоматическая (8 больных).
Болезнь Вильсона-Коновалова встречается одинаково часто как у женщин, так и у мужчин. В последние годы наблюдается тенденция к увеличению числа диагностируемых случаев. Распространенность заболевания в среднем составляет 30 случаев на 1 млн. человек. Высокая заболеваемость отмечается в регионах, где существуют близкородственные браки (Иран, Йемен, Ирландия), а также в Японии и на острове Сардиния. Так, в Японии болезнь Вильсона-Коновалова диагностируется с частотой 1:30 тысяч; а для сравнения в Австралии — 1:100 тысяч населения. Из-за увеличения случаев болезни Вильсона-Коновалова необходимо проявлять большую диагностическую настороженность, особенно в местах с частым проявлением этого заболевания.
Гепатолентикулярная дегенерация является причиной 15—20 % всех заболеваний печени у детей. Зная основные признаки заболевания и ее генетические аспекты, можно предостеречь болезнь на ранних стадиях. При любом подозрении на болезнь Вильсона-Коновалова нужно обязательно делать прямую или косвенную ДНК — диагностику с целью уточнения диагноза. При проведении прямой ДНК — диагностики необходимо проводить первоочередной поиск мутаций в гене АТР7В, основываясь на данных о частоте мутаций, встречающихся на данной местности. При выявлении у пациентов с предположительным диагнозом болезни Вильсона-Коновалова, мутаций в гене АТР7В на обеих хромосомах (доклиническая стадия), необходимо начинать превентивную патогенетическую терапию.
Раннее выявление заболевания — ключ к быстрому выздоровлению!
Список литературы:
1. Автореферат диссертации на соискание учебной степени кандидата медицинских наук «Болезнь Вильсона у детей: диагностика, течение и прогноз», Четкина Т.С., Москва, 25.08.2011 год.
2. Автореферат диссертации на соискание ученой степени кандидата медицинских наук «Гено-фенотипические корреляции при болезни Вильсона в Республике Башкортостан», Магжанова А.Р., Уфа, 31.05.2007 год.
3. Автореферат диссертации на соискание учебной степени кандидата медицинских наук Г.Я. Левиной, «Морфологические изменения в печени при гепато-церебральной дистрофии (болезни Вильсона-Коновалова)», Москва, 1968 год.
4. Canadian liver foundation, Wilson disease, Canada, 2014 год.
5. Centre Hépato-Biliaire — Hôpital Universitaire Paul Brousse, la maladie de Wilson, France, 08.10.2014 год.
6. Polymorphism of neuropsychiatric disorders of Wilson disease's patients in Volga-Ural population / A. Magzhanova, A. Karunas, R. Magzhanov, E. Khusnutdinova, “World Congress of Psychiatric Genetics October — Boston”, 2005 год.
