Статья:




ДИАГНОСТИКА И ЛЕЧЕНИЕ ПАЦИЕНТОВ С СИНДРОМОМ МАРФАНА
Секция: 2. Биологические науки
Выходные данные
Насонов А.Д. ДИАГНОСТИКА И ЛЕЧЕНИЕ ПАЦИЕНТОВ С СИНДРОМОМ МАРФАНА // Молодежный научный форум: Естественные и медицинские науки: электр. сб. ст. по мат. XXIII междунар. студ. науч.-практ. конф. № 4(22). URL: https://nauchforum.ru/archive/MNF_nature/4(22).pdf (дата обращения: 30.07.2026)
Лауреаты определены. Конференция завершена
Эта статья набрала 4087 голосов
Мне нравится4087
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников

XXIII Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки»
ДИАГНОСТИКА И ЛЕЧЕНИЕ ПАЦИЕНТОВ С СИНДРОМОМ МАРФАНА
Насонов Алексей Дмитриевич
студент, ПМГМУ им. И.М. Сеченова, РФ, г. Москва
Аракелян Валерий Сергеевич
научный руководитель, проф., д-р мед. наук, руководитель отделения хирургии артериальных патологий Научного центра сердечно-сосудистой хирургии им. А.Н. Бакулева РАМН, РФ, г. Москва
Ларина Светлана Николаевна
ВВЕДЕНИЕ.
Синдром (болезень) Марфана(СМ) — это врожденное нарушение строения соединительных тканей тела с характерным поражением ряда систем организма (костно-мышечной, сердечно-сосудистой, респираторной, центральной нервной) и глаз. Актуальностью проблемы при Синдроме Марфана была и остается нарушение сердечно сосудистой системы. Отсутствие наблюдения за такими пациентами и обособленная тяжесть всего заболевания приводят к расслоению, или полному разрыву аорты, в последствие приводящие к летальному исходу. В первую очередь важна диагностика и наблюдение за такими пациентами с самого раннего периода заболевания, а при поздних стадиях — хирургическое лечение
Историческая Справка.
В 1896 г. A. Marfan наблюдал аномалию скелета у 5-летней девочки, проявляющуюся арахнодактилией (необыкновенно длинные и тонкие конечности с удлинением пальцев рук и ног), врожденными контрактурами и сколиозом. Чуть позже были описаны случаи эктопии хрусталиков, нарушения соединительной ткани и лишь в 1943 г. R. Baer, H. Taussing и E. Oppenheimer впервые отметили нарушения сердечно-сосудистой системы, наблюдаемые у больных с СМ. Авторы описали гистопатологические изменения в стенке аорты, которые являлись причиной образования аневризм.
Наиболее полные исследования СМ были проведены к 1972 г. V. McKusick (американский генетик), который собрал самую большую серию больных и описал широкий диапазон нарушений сердечно-сосудистой системы при СМ и их последствия. С середины 50-х годов генетическая клиника Moore в Johns Hopkins Hospital ведет исследования этих больных и обладает наибольшим материалом, касающимся больных с СМ [3].
Этиология (генетические предпосылки развития синдрома Марфана).
Синдром Марфана — наиболее частое генетическое (аутосомно-доминантное) нарушение у людей, которое встречается с частотой 1:10000 во всех географических регионах и этнических группах. Примерно в 75 % случаев заболевание передается генетически и только 25 % вызываются спорадическими мутациями. Этот синдром связан с дефектом 15-й хромосомы, отвечающая за информацию о структуре фибриллина. Фибриллин представляет из себя белок микрофибрилл, который вместе с эластином является основной частью системы эластических волокон.
СМ, как уже отмечено, обусловлен мутациями в гене, кодирующем основной составной компонент микрофибрилл — фибриллин-1 (FBN1) [5]. Микрофибриллы формируют основу, на которой располагается эластин. Комплексы микрофибрилла с эластином представляют собой эластические волокна. Именно дефектом микрофибрилл специалисты объясняют разнообразные проявления СМ [3].
Симптомы и диагностические критерии Синдрома Марфана.
На сегодняшний момент диагностические критерии базируются на больших и малых клинических симптомах, которые были установлены в 1996 г. (Ghent Criteria) [6].
Большие критерии состоят из триады симптомов включающие в себя нарушения сердечно-сосудистой системы, опорно-двигательного аппарата и нарушение функции зрения Малые критерии включают дополнительные патологические признаки характерные к выще указанным органам и системам, а также специфические нарушения дыхательной системы, кожи.
Исходя из этих данных подтверждение Синдрома Марфана основывается на обнаружение у пациента 2 больших критерий и тех или иных признаков малого критерия. Если же в семье ранее подтверждалось поражение гена, то для установления диагноза достаточно одного большого критерия.
Поражение мышечно-костной системы.
Изменения скелета отмечают в ⅔ пациентов с СМ, и включают: высокий рост, астеническую телосложение, долихостеномелия (увеличенная длина нижних конечностей по отношению к туловищу), прогнатию, воронкообразную грудную клетку удлиненные пальцы рук с узкими ногтевыми пластинами (арахнодактилия — встречается у 77 % пациентов), сколиоз (у 72 % женщин и 50 % мужчин), кифосколиозы, нарушение функции суставов (связанны с ослаблением связок, приводящие к гипермобильности суставов кистей и стоп), плоскостопие [3].
Нарушение функций глаз и зрения.
Глазные симптомы выявляют у 80 % мужчин и 60 % женщин с СМ. Обычно у пациентов с СМ наблюдаются близорукость, но и в некоторых случаях фиксируется дальнозоркость. Наиболее характерное нарушение функции зрения это — подвывих хрусталика глаза (встречается у 77 % пациентов). При Синдроме Марфана расположение хрусталика преимущественно имеет неординарный характер (вверх и наружу). С возрастом почти у всех пациентов наблюдается катаракта. А в некоторых случаев в возрастном периоде возникает отслоение сетчатки [3].
Поражение сердечно сосудистой системы.
Наиболее важным и серьезным признаком Синдрома Марфана, это нарушение сердечно сосудистой системы. Примерно у 1/3 больных имеется какой-либо врожденный порок сердца. Частыми осложнениями со стороны сердечно-сосудистой системы являются пролапс митрального клапана и расширение аорты. В детском возрасте чаще поражается митральный клапан, в то время как юношеские годы и у взрослых людей встречается недостаточность аортального клапана Аневризма аорты при СМ возникает с одинаковой частотой у мужчин и женщин в возрасте 30—40 лет, с преимущественным поражением восходящей части, имеет мешковидный вид, с характерным поражением ее ветвей. Аневризмы могут возникать не только в разных отделах аорты, но и в легочной артерии, а также в сонных, лучевых, локтевых, бедренных и других сосудах организма [1]. Аневризма аорты при СМ опасна не только возможностью разрыва или стенозом коронарных артерий, но и развитием нарушений мозгового кровообращения, что может привести к инвалидности или смерти пациентов с СМ [3].
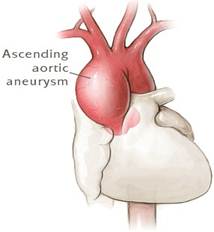
Рисунок 1. Аневризма восходящей аорты
Естественное течение и статистика.
Средняя продолжительность жизни при СМ составляет от 30 до 50 лет. Основные причины летальности — осложнения сердечно-сосудистой системы, причем дилатация аорты при этом наблюдается в 60—80 % случаев. Разрыв и расслоение аорты наряду с сердечной недостаточностью клапанного генеза — наиболее частые причины гибели больных. Митральная недостаточность клапанов сердца является основной причиной гибели детей с СМ.
В статистике Methodist Hospital касающейся 280 операций:
· 151 пациентов — протезирование аортального клапана в 135 случаях;
· 13 пациентов — протезирование митрального клапана;
· 10 пациентов — протезирование аорты на всем протяжение, путем поэтапных вмешательств;
· 26 пациентов — выполнялась протезирование почти всей аорты;
· 7 пациентов — грудной аорты.
Расслоение аорты отмечено у 102 (67 %) пациентов, из них у 61 пациентов — она локализовалось в восходящей аорте (60 %).
Из всей группы больных 105 (70 %) имели аортальную регургитацию, 76 (50 %) — пролапс митрального клапана; 29 (20 %) — недостаточность митрального клапана.
Характерно, что 34 больных (23 %) были оперированы повторно из-за того, что во время первичной операции производилось раздельное протезирование аортального клапана и восходящей аорты, или из-за осложнений, связанных с применявшейся ранее оперативной техникой.
В результате проведенного лечения 30-дневная выживаемость составила — 94 %, а 5 — и 10-летняя — 75 и 50 % соответственно [3].
ЛЕЧЕНИЕ.
Медикаментозная терапия должна проводиться как до, так и после операции. Целью лечения должно быть строгий контроль артериального давления (систолическое давление до 120 мм рт. ст. Для пациентов с расслоением аорты — 110 мм рт. ст.). Чаще всего применяют блокаторы бета-адренорецепторов (бизопролол), которые замедляют развитие или прогрессирование расслоения аорты. Блокаторы рецепторов ангиотензина II является потенциально полезными, поскольку они приводят к TGF-beta-антагонизма. Современные клинические испытания демонстрируют высокую эффективность лозартана для профилактики развития аневризмы аорты у пациентов с СМ [7].
Клиническое течение заболевания.
В данной работе рассмотрен опыт хирургического лечения пациентов с СМ. У пациента с поражением грудного и брюшного отдела аорты на основе проведенного анализа непосредственных результатов, выявлена клиническая эффективность выполнения хирургического вмешательства. Клиническое обследование проводили на госпитальном этапе, включая анализ данных анамнеза, клинических и инструментальных методов исследования.
Данный клинический случай является иллюстрацией удачного хирургического лечения пациента возрастом 23 лет.
Клиническое наблюдение.
Пациент К. поступил на лечение в ОАП НЦССХ РАМН в возрасте 23 лет с жалобами на наличие пульсирующего образования в животе.
Анамнез заболевания: В 10 лет установлен Синдром Марфана (генетическое подтверждение, фенотипические проявления), рекомендаций не получено, АД не измерял, работал грузчиком. В течение 1 года отмечает наличие пульсирующего образования в животе. В сентябре 2013 года получал лечение по поводу ожога правой кисти и голени, тогда же впервые обследован (УЗИ брюшной полости), заподозрена аневризма брюшной аорты и при дообследовании (МСКТ-АГ) выявлена коарктация аорты и РА 3 типа с сформировавшейся аневризмой всей нисходящей аорты и обеих общих подвздошных артерий. Поступил на оперативное лечение.
Цифры артериального давления максимально 170/100, привычно 130/80. Характер течения артериальной гипертензии: до сентября 2013 года АД не измерял.
Объективное обследование.
Общее состояние удовлетворительное. Сознание ясное. Активность активен. Конституционные особенности нормостеник.
Рост (см) = 187. Вес (кг) = 69,4. Индекс массы тела = 19,85. Строение тела неправильное деформации, характерные синдрому Марфана (сколиоз, арахнодактилия, миопия). Развитие подкожной клетчатки снижено. Поверхностные послеожоговые рубцы на правой кисти и голени.
Дыхательная система: Грудная клетка воронкообразная. Частота дыхательных движений 16 в мин. Дыхание жёсткое, проводится во все отделы. Хрипы нет. Данные перкуссии: ясный легочный звук. Сердечно-сосудистая система. Тоны сердца ясные, ритмичные. Шумы сердца: систолический по 2-ом м/р слева, в межлопаточном пространстве. ЧСС = 68 уд./мин. АД: на правой руке — 120/80 мм рт. ст., одинаково на обеих руках. Пульс удовлетворительного наполнения = 68 уд./мин., пульсация БЦА отчетливая, симметричная, артерий н/к ослаблена, сохранена на всех уровнях. Аппетит не нарушен. Печень не увеличена. Селезенка не пальпируется. Живот мягкий, в области мезогастрия определяется объемное пульсирующее образование плотно-эластической консистенции d около 8 см, безболезненное при пальпации. Стул регулярный. Мочеотделение свободное, безболезненное
ЭКГ: ЭКГ (13.03.2014): Ритм сердца синусовый. Частота сердцебиений в минуту=50. Положение электрической оси сердца: нормальное.
Длина интервала: PQ=0,16 сек. QRS=0,09 сек. QRST=0,4 сек. Блокада пнпГ. Гипертрофия миокарда ЛЖ.
КТ, Ядерн.диагн.: МРТ ГМ (04.03.2014):
МСКТ-АГ (14.10.2013): восходящая и дуга аорты без особенностей, d восходящей аорты 36, дуги — 34 — 26 мм; на 15 мм дистальнее устья левой подключичной артерии — коарктация аорты с сужением просвета аорты на 25 %, дистальнее — расслоение аорты до бифуркации аорты, пристеночные тромбы в ложном просвете аневризма аорты на всем протяжении с переходом на обе ОПА, максимальный диаметр аневризмы 80 мм, ЧС и ВБА отходят от истинного просвета, обе почечные артерии от ложного просвета.
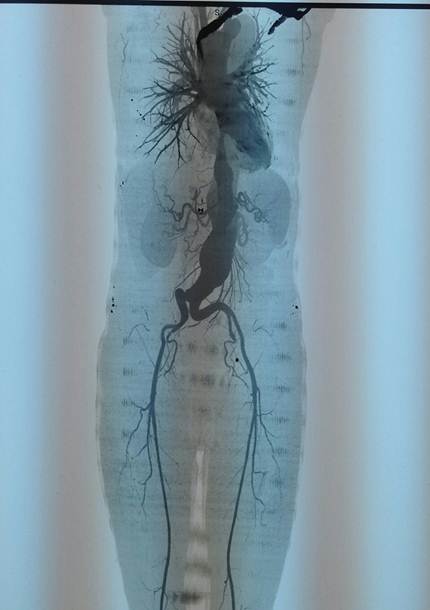
Рисунок 2. КТ — АГ аорты А) Срединная (сагиттальная) проекция
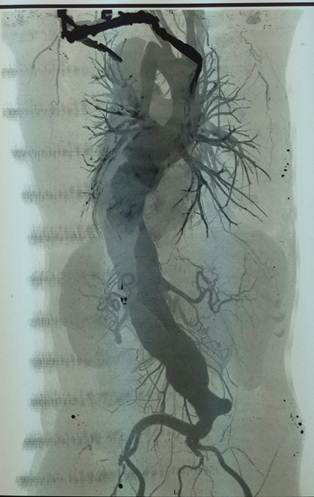
Рисунок 3. КТ — АГ аорты Б) Фронтальная проекция
Учитывая прогрессирование течения заболевания, высокий риск осложнений, связанных с артериальной гипертензией, характер поражения нисходящей аорты, ее диаметр, было принято решение о проведении хирургического вмешательства резекции аневризмы грудной и брюшной аорты с протезированием
Диагноз до операции.
Синдром Марфана: коарктация аорты, неполная форма. Расслаивающая аневризма аорты 3 типа с отхождением почечных артерий от ложного просвета. Аневризма инфраренального отдела аорты и левой общей подвздошной артерии.
Название операции: Резекция расслаивающей аневризмы аорты в грудном и брюшном отделах с бифуркационным протезированием тканым эксплантатом 22—11 мм с пластикой висцеральных и почечных артерий, реконструкцией критических межреберных и поясничных артерий, в условиях ИК.
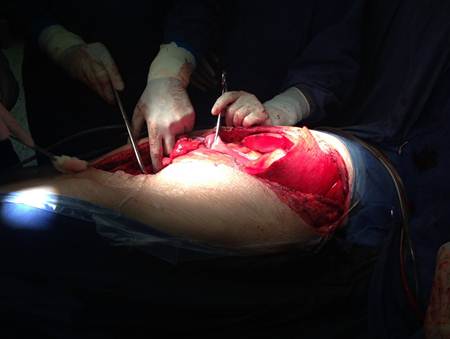
Рисунок 4. Доступ к аневризме был выполнен с помощью торакофренолюмботомии. Послойно рассечены мягкие ткани до забрюшинного пространства и выделены подвздошные артерии
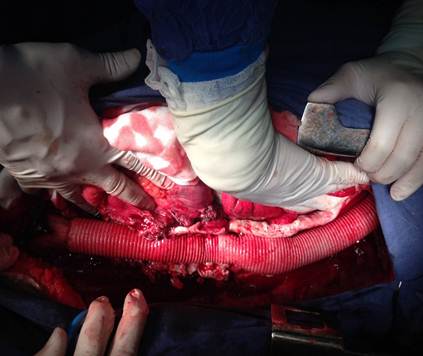
Рисунок 5. Спротезированный участок грудного и брюшного отдела аорты тканевым эксплантатом. Спротезированы подвздошные артерии. Почечные и висцеральные артерии имплантированы в протез
К концу первых суток после операции больной в удовлетворительном состоянии был переведен из ОРИТ в палату отделения.
На 10 сутки после операции состояние пациента удовлетворительное. Дыхание жесткое проводится во все отделы легкого, хрипов нет. Тоны сердца ясные, ритмичные, шумов нет. Артериальное давление 130/90 мм рт. ст. ЧСС 78 уд./мин. Печень не увеличена.
Ранний послеоперационный период протекал без осложнений, рана зажила первичным натяжением. Медикаментозная терапия: анальгетики, антибиотики, НПВП.
ЗАКЛЮЧЕНИЕ.
Обсуждение.
В описанном нами случае причиной грудной и брюшной аневризмы аорты стали несколько факторов: генетическая предрасположенность, связанная с Синдромом Марфана, недостаточные терапевтические рекомендаций по лечению СМ.
Показанием к операции явилась выраженная артериальная гипертензия рефрактерная к лекарственной терапии (среднее систолическое АД составляло более 160 мм рт. ст., при возрастной норме до 120 мм рт. ст.).
Также показанием к хирургическому лечению стало диаметр аорты 80 мм.
В настоящее время хирургическим методом лечения торакоабдоменнальных аневризм в случаях пациентов с СМ является протезирование пораженных участков аорты (протезирование аорты тканевым эксплантатом).
Нами был описан случай радикального хирургического лечения 23-летнего пациента с гипоплазией всей нисходящей грудной аорты, которая была выполнена с хорошими непосредственными результатами.
Синдром Марфана на сегодняшний день остается тяжелым генетическим заболеванием, и является трудно диагностируемым. Благодаря хирургическому лечению таких пациентов останавливается рост аорты и ее расслоение. Результатом таких вмешательств сказывается на общем улучшение состояния пациента, снижается артериальное давление (до 170/100 после 130/80) тем самым облегчая нагрузку сердца. Также уменьшается диаметр аорты (до 8 см после 2,5—3 см). Не стоит забывать, что СМ — это заболевания которое остается зависимым от хирургии, так как не какие инноваций в лечение сосудистых заболеваниях на данный момент не способны вылечить симптомы этого синдрома (дилатация аорты, дефекты клапанов сердца).
В завершение данной работы следует отметить следующее. Несмотря на то что СМ представляет собой очень серьезную патологию и прогноз при наличии такого грозного проявления заболевания, как аневризма восходящей аорты, казалось бы, должен быть неблагоприятным, современные хирургические и терапевтические методы и схемы лечения позволили пролонгировать среднюю продолжительность жизни пациентов с СМ с 48 до 61—72 лет.
Список литературы:
1. Ватутин Н.Т., Склянная Е.В., Кетинг Е.В. (2006) Синдром Марфана. Кардиология, 1: 92—98.
2. Лисиченко А.В. (1986) Синдром Марфана. Наука, Новосибирск, 164 с.
3. Покровский А.В. — Клиническая ангиология. Том 1 С. 490—560.
4. Смоленский В.С., 1964; Белоконь Н.А., Кубергер М.Б., 1987.
5. Фищенко Я.В. (2006) Алгоритм диагностических проявлений синдрома Марфана.
6. De Pape A., Devereux R.B., Ditz H.C. et al. (1996) Revised diagnostic criteria for the Marian syndrome Am. J. Med. Genet. 62(4) 417—426.
7. Karalliedde J., Viberti G. Microalbuminuria and cardiovascular risk. Am J Hypertens, 2004; 17: 986—93.
