Статья:




МИТОХОНДРИАЛЬНЫЕ ЗАБОЛЕВАНИЯ
Секция: 2. Биологические науки
Выходные данные
Киракосян Е.В. МИТОХОНДРИАЛЬНЫЕ ЗАБОЛЕВАНИЯ // Молодежный научный форум: Естественные и медицинские науки: электр. сб. ст. по мат. XXIV междунар. студ. науч.-практ. конф. № 5(23). URL: https://nauchforum.ru/archive/MNF_nature/5(23).pdf (дата обращения: 29.07.2026)
Лауреаты определены. Конференция завершена
Эта статья набрала 52 голоса
Мне нравится52
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников

XXIV Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки»
МИТОХОНДРИАЛЬНЫЕ ЗАБОЛЕВАНИЯ
Киракосян Евгения Валериковна
студент лечебного факультета Первого МГМУ им. И.М. Сеченова РФ, г. Москва
Филиппова Алла Викторовна
научный руководитель, канд. мед. наук, доц. кафедры биологии и общей генетики Первого МГМУ им. И.М. Сеченова, РФ, г. Москва
Дегтяревская Татьяна Юрьевна
Введение.
Митохондрии — это энергетические станции клетки, которые встречаются почти в каждой клетке человеческого организма. Они удивительны тем, что имеют свою собственную кольцевую молекулу ДНК. Результаты исследований последних лет показывают, что мутации митохондриальной ДНК могут стать причиной развития нескольких типов заболеваний.
Митохондрии — небольшие органеллы клетки, окруженные двойной мембраной и, как было отмечено, имеющие свою кольцевую молекулу ДНК. Размер митохондрий составляет 0,5—1 мкм. Вот так выглядит митохондрия на снимке электронного микроскопа:
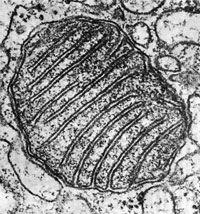
Рисунок 1. Митохондрия на снимке электронного микроскопа
Строение митохондрии.
Митохондрии состоят из наружной мембраны, межмембранного пространства и внутренней мембраны, которая образует выпячивания — кристы, вдающиеся в матрикс. Матрикс заполняет пространство, окруженное внутренней мембраной митохондрии.
Внешняя мембрана митохондрий свободно проницаема для воды и ионов, поэтому ионный состав межмембранного пространства практически не отличается от цитозоля (раствора, заполняющего клетку). Однако белки не могут свободно проникать через внешнюю мембрану митохондрий и должны иметь специальные — сигнальные — последовательности, чтобы попасть внутрь митохондрии. В межмембранном пространстве митохондрии постоянно находится цитохром С.
На внутренней мембране митохондрии находятся белки, участвующие в цепи окислительного фосфорилирования, а также АТФ-синтаза, которая синтезирует АТФ. Митохондрии часто называют «энергетическими станциями клетки», поскольку одна из их главных функций — это выработка энергии.
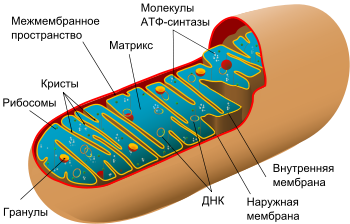
Рисунок 2. Схема строения митохондрии
Митохондрии имеют свою собственную, митохондриальную, ДНК. Конечно, размер последовательности ДНК, заключенной в митохондриях, гораздо меньше той ДНК, которая компактно уложена в хромосомы в ядре. Тем не менее, мутации митохондриальной ДНК могут стать причиной развития как минимум нескольких типов заболеваний.
В организме человека митохондрии — это единственное место нахождения внеядерной ДНК. Структура и функции митохондрии находятся под двойным генетическим контролем. Что это значит? Дело в том, что часть белков, работающих в митохондриях, синтезируется на основе генетической информации, закодированной в ядерной ДНК. Митохондриальная же ДНК, состоящая из 37 генов, кодирует преимущественно аминокислотные последовательности белков — участников дыхательной цепи, а также информацию для молекул РНК, участвующих в транскрипции и трансляции митохондриальной ДНК.
Интересно, что белки, отвечающие за репликацию митохондриальной ДНК, синтезируются на матрице хромосомной ДНК, поэтому иногда сложно понять, почему происходит та или иная мутация митохондриальной ДНК.
Количество митохондрий в клетке зависит от ткани, к которой клетка принадлежит, и от энергетических потребностей. Митохондрии могут делиться (процесс, похожий на деление бактериальной клетки) при увеличении потребности клетки в энергии. При снижении потребностей клетки в энергии митохондрии разрушаются.
Так выглядит деление митохондрий под электронным микроскопом:
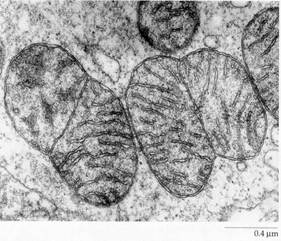
Рисунок 3. Деление митохондрий под электронным микроскопом
У человека митохондриальная ДНК, как и сами митохондрии, как правило, наследуется лишь от женской половой клетки, то есть передается только по материнской линии. Митохондрии, которые приносит сперматозоид, разрушаются и не влияют впоследствии на работу клеток развивающегося организма. Заболевания, связанные с мутациями митохондриальной ДНК, как правило, вариабельны, и диагностировать их сложно. Однако наряду с этим накапливаются факты, указывающие на участие митохондриальных мутаций в старении организма, в развитии диабета и онкологических заболеваний.
Митохондриальная генетика отличается от менделевской по ряду признаков. Например, в митохондриальной ДНК имеется множество копий одной и той же последовательности, что приводит к интересному результату. Если мутация встречается во всех копиях митохондриальной ДНК (гомоплазмическая мутация), она, как правило, проявится в фенотипе. Если же мутация представлена лишь в нескольких копиях ДНК (гетероплазмическая мутация), то в фенотипе она проявится лишь при определенном — критическом — уровне встречаемости этой мутации в копиях последовательности ДНК. Таким образом, для митохондриальных мутаций есть порог, лишь после преодоления которого они встречаются в фенотипе.
Исследования показали, что риск развития заболевания у детей матери, имеющей единичную мутацию митохондриальной ДНК, составляет 4,11 %. Кроме того, гомоплазмические мутации передаются потомкам, но могут не проявить себя в виде заболевания. Так, у детей пациенток с врожденной нейропатией зрительного нерва Лебера лишь 50 % сыновей, но 90 % дочерей страдают аналогичным заболеванием. Вместе с тем некоторые мутации проявятся в фенотипе только при действии факторов среды – например, мутация RNR1, ведущая к глухоте, проявится в фенотипе лишь при назначении аминогликозидных антибиотиков.
Характеристика митохондриальных заболеваний.
Митохондрии обеспечивают жизнедеятельность любой клетки, поэтому неудивительно, что митохондриальные заболевания поражают многие ткани и органы организма. Из-за этого бывает сложно оценить вклад именно митохондриальных мутаций в развитие заболевания, однако совокупность признаков поражения многих систем органов помогает поставить правильный диагноз.
Впервые нарушения митохондриального генома были описаны в 1988 году после изучения двух классических заболеваний — синдрома Кирнса-Сэйра (Kearns-Sayre) и врожденной нейропатии зрительной нерва Лебера (Leber’s hereditary optic neuropathy).
Синдром Кирнса — Сэйра — это редкое неврологическое заболевание, которое развивается, как правило, в возрасте до 20 лет. Синдром характеризуется прогрессирующим снижением функции глазодвигательных мышц, вплоть до полного их паралича. Эти симптомы часто сопровождаются слабостью мышц века и его опущением (птоз). Дополнительными симптомами заболевания могут быть небольшая слабость мышц туловища и конечностей, нарушения сердечной проводимости, потеря слуха, неспособность производить координированные движения (атаксия), а также диабет.
Врожденная нейропатия зрительного нерва Лебера развивается из-за гибели ганглионарных клеток сетчатки и их отростков, что ведет к снижению остроты зрения. Заболевание чаще поражает мужчин и проявляется в молодом возрасте. Как правило, в начале поражается один глаз, затем второй. У здорового человека диск зрительного нерва выглядит светло-желтым, с четкими краями. А вот так выглядит глазное дно при нейропатии Лебера — отечный диск зрительного нерва с размытыми краями:
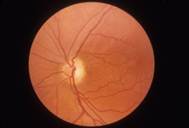
Рисунок 4. Глазное дно при нейропатии Лебера — отечный диск зрительного нерва с размытыми краями
В последнее время офтальмоплегию, синдром Пирсона, синдром Лея, рабдомиолиз стали связывать с мутациями митохондриальной ДНК.
Синдром Пирсона (Pearson syndrome) характеризуется сидеробластной анемией и дисфункцией поджелудочной железы — как ее экзокринного аппарата, что проявляется недостаточной выработкой пищеварительных ферментов, так и эндокринного, что ведет к нарушению выработки инсулина и диабету.
Это заболевание проявляется уже в детском возрасте и часто ведет к летальному исходу. К описанным симптомам часто добавляется и синдром Кирнса-Сэйра.
Синдром Лея (Leigh’s syndrome) — это крайне редкое заболевание, которое поражает прежде всего центральную нервную систему. Заболевание проявляется в детском возрасте и характеризуется потерей мышечного тонуса, судорогами, нарушением роста и развития ребенка, нарушением функций почек.
Рабдомиолиз — заболевание, связанное с разрушением клеток мышечной ткани — миоцитов, сопровождающееся снижением силы мышц и ограничением движений — прежде всего конечностей.
Заключение.
Настоящую сложность для врача-клинициста представляет задача выявления участия мутаций митохондриальной ДНК в развитии более «обыденных» заболеваний. Например, митохондриальные аномалии называют и среди причин развития сахарного диабета. Кроме того, было показано, что определенная гомоплазмическая мутация часто встречается при метаболическом синдроме, который включает в себя гипертензию, гиперхолестеролемию и гипермагниемию. Хотя, конечно, необходимо принимать во внимание тот факт, что подобного рода мутации описываются лишь для конкретных семей, а не для популяции в целом.
Список литературы:
1. Greaves L., Reeve A., Taylor R., Turnbull D. Mitochondrial DNA and disease. Journal of pathology, 2012.
2. Park C.B., Larsson N.G. Mitochondrial DNA mutations and aging. Journal of cell biology, 2011.
