Статья:




ГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ СИНДРОМА АНГЕЛЬМАНА И ИХ ФЕНОТИПИЧЕСКИЕ ПРОЯВЛЕНИЯ
Секция: 4. Медицинские науки
Выходные данные
Отрошко Е.В. ГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ СИНДРОМА АНГЕЛЬМАНА И ИХ ФЕНОТИПИЧЕСКИЕ ПРОЯВЛЕНИЯ // Молодежный научный форум: Естественные и медицинские науки: электр. сб. ст. по мат. XXIV междунар. студ. науч.-практ. конф. № 5(23). URL: https://nauchforum.ru/archive/MNF_nature/5(23).pdf (дата обращения: 27.07.2026)
Лауреаты определены. Конференция завершена
Эта статья набрала 46 голосов
Мне нравится46
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников

XXIV Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки»
ГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ СИНДРОМА АНГЕЛЬМАНА И ИХ ФЕНОТИПИЧЕСКИЕ ПРОЯВЛЕНИЯ
Отрошко Елена Вячеславовна
студент Первого МГМУ им. И.М. Сеченова, РФ, г. Москва
Молодожникова Наталья Михайловна
научный руководитель, канд. биол. наук, доц. кафедры биологии и общей генетики Первого МГМУ им. И.М. Сеченова, РФ, г. Москва
Введение.
Синдром Ангельмана (далее СА) — генетическая аномалия, о механизмах возникновения которой и пойдет речь в этой статье. Синдром Ангельмана обычно не диагностируется в раннем детстве, так как дефекты в развитии являются в это время неспецифическими. Наиболее распространенный возраст диагностики составляет от 2 до 5 лет, когда поведенческие характеристики становятся более очевидны. Дети могут иметь относительно широкий рот и высунутый язык, иногда встречается выдающийся подбородок (см. рисунок). Существует гипотеза, что фенотипическое проявление СА зависит от механизма генетического нарушения. Данные об этом будут приведены ниже. Синдром Ангельмана является хорошо определяемым клиническим состоянием, в основном из-за характерного поведения и хода развития.
 Рисунок 1. Дети с синдромом Ангельмана
Рисунок 1. Дети с синдромом Ангельмана
Возрастные и физические данные.
Постоянные (100 %): Функционально выраженные задержки развития; расстройства движения и равновесия, обычно атаксия или тремор конечностей; нарушение координации может быть слабым, или проявиться не как открытая атаксия, а как качание, неустойчивость, неуклюжесть, или быстрые, отрывистые движения;поведенческая уникальность: любое сочетание частых движений — хлопает или машет руками, смеется, улыбается, кажется счастливым, легко возбудим, часто гиперактивен; нарушения речи: исключение, или сведение к минимуму использование слов, восприятие и навык невербальных коммуникаций выше, чем вербальных
Частые (более 80 %): Задерживается рост черепа, что приводит к микроэнцефалии в возрасте 2 лет. Микроэнцефалия является более выраженной у лиц с 15q11.2-q13делецией.Судороги обычно начинаются до трехлетнего возраста. Выраженность судорожных припадков, как правило, уменьшается с возрастом, но они продолжаются на протяжении всей жизни.Аномальные ЭЭГ, с характерным рисунком. ЭЭГ-аномалии могут возникать в первые 2 года жизни и предшествовать клиническим признакам, часто не коррелируют с клинической картиной событий.
Сопутствующие (20—80 %): Плоский затылок, затылочная борозда, высунутый язык, расстройства движений языка, сосания, глотания. Проблемы с кормлением в младенчестве; широкий рот, широко расставленные зубы. Частое чрезмерное слюноотделение; Чрезмерные жевательные движения; Косоглазие. Слабая пигментация кожи, светлые волосы и цвет глаз (по сравнению с семьей); Гиперактивность нижних конечностей, глубокие сухожильные рефлексы; Поднятые руки во время вставания; Широкие шаги с вывернутыми ногами; Повышенная чувствительность к жаре; Необычные циклы сна и бодрствования, уменьшенная потребность во сне; Увлечение водой и складчатыми предметами, такими как некоторые виды бумаги или пластмасс; Необычное пищевое поведение; Ожирение (у детей в старшем возрасте); Сколиоз; Запор;
Генетические механизмы, вызывающие СА.
В 1997 году, мутации в гене, расположенном на хромосоме UBE3A 15, были идентифицированы как причина синдрома Ангельмана [7, с. 3—70; 10, с. 7—74]. Все известные механизмы возникновения СА — нарушение, инактивация или отсутствие этого гена на материнской 15 хромосоме. Существует несколько генетических «классов» или механизмов, которые могут нарушить UBЕ3A и таким образом вызвать СА [6, с. 1—6; 9, с. 73—1867]. Эти механизмы изображены на этом рисунке.
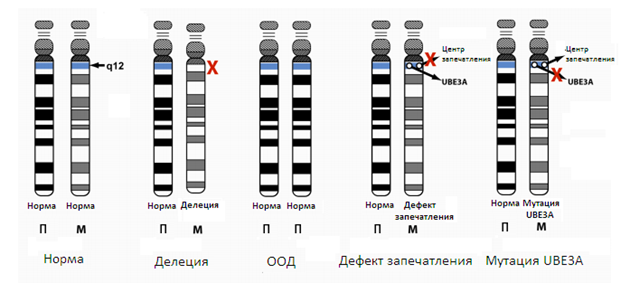 Рисунок 2. Механизмы возникновения синдрома Ангельмана
Рисунок 2. Механизмы возникновения синдрома Ангельмана
Показана 15 пара хромосом для каждого механизма, стандартный набор хромосом изображен слева с нормальной В12 хромосомной областью. П =отцовская хромосома и М = материнская. САможет быть вызван обширной делецией области 15q12 области материнской хромосомы (где расположен активатор UBЕ3A гена). А также может быть вызван наследованием от отца 2 отцовской хромосомы; явление называется отцовская однородительская дисомия (ООД). Еще одной причиной может быть дефект запечатления(ИД), который возникает, когда 15 хромосома, унаследованная матерью по отцовской линии, функционирует так, что экспрессияUBЕ3A фактически выключена. Центр запечатления находится на некотором расстоянии от UBЕ3A, но он способен регулировать его посредством сложных механизмов, что является предметом интенсивных исследований. Наконец, СА может быть вызван мутацией в гене UBЕ3A материнской 15 хромосомы.
Таблица 1.
Распространенность механизмов
Механизм |
Частота (%) |
Делеция |
~70 |
ООД |
2—3 |
Дефект запечатления |
3—5 |
Мутация UBE3A |
5—10 |
Другие хромосомные нарушения |
1—2 |
Неизвестно |
10—15 |
Таблица показывает распространенность каждого генетического механизма, а также отмечает, что около 10—15 % людей с клиническими характеристиками СА имеют нормальные генетические показатели. В этом случае неясно имеют ли эти лица правильные диагнозы или другие неидентифицированные генетические дефекты, которые вызывают СА.
Наиболее частой генетической причиной, приводящей к СА, является делеция. На схеме изображенаболее подробная информация об этом. Типичная делеция региона является действительно обширной и охватывает около 6 миллионов молекул (пар оснований) ДНК. Большинство делеций происходят в одной точке разрыва (ВР1) либо ВР2 или ВР3 и называются I или II класс делеции. Около 10 % делеций распространяются за пределы ВР3, например, на сайт BP4. Новые методы клинических испытаний, например, на основе массива сравнительной геномной гибридизации могут различать I и II класс делеций. Однако FISH тест не может этого определить. Все обширные делеции удаляют UBЕ3A на материнской хромосоме. Делеция также затрагивает соседние гены, как на фото (например, гены ГАМК рецепторов), но делеция UBЕ3A является причиной практически всех проблем, связанных с СА.
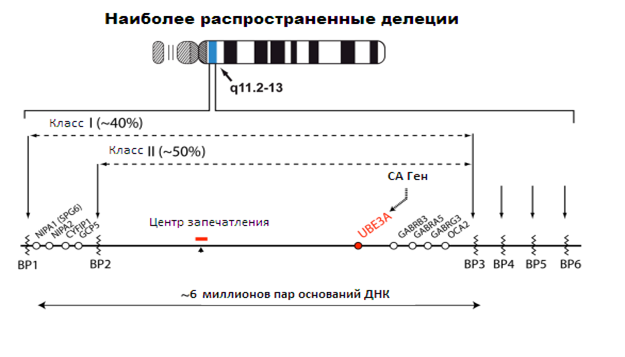 Рисунок 3. Наиболее распространенные делеции
Рисунок 3. Наиболее распространенные делеции
UBЕ3A и пути распространения Убиквитина.
UBЕ3A ген производит белок UBE3A (также называемый Е6-АР) и этот белок является важным компонентом пути образования убиквитин — протеосом (на рисунке ниже). Этот путь крайне важен для всех клеток, особенно нейронов головного мозга. Он позволяет маленькой белковой молекуле убиквитина прикрепиться к определенному белку, тем самым заставляя их взаимодействовать [13, с. 3—81]. Убиквитин — небольшой белок (76 аминокислот в длину), который может маркировать другие белки в целях инициации их уничтожения. На рисунке показано, как Elи Е2 активируют и передают убиквитин на Е3. Существует множество типов Е3 и UBЕ3A является одним из них. UBЕ3A способен химически присоединить убиквитин к белкам-мишеням. Важно, что вструктуре UBЕ3A находится НЕСТ домен, молекулярный карман, который позволяет убиквитину и белку-мишени сблизиться, чтобы прикрепить активированный убиквитин [14, с. 249—59]. Некоторые белки-мишени UBЕ3A известны, но в настоящее время неизвестно, какой из белков-мишеней связан с мозговыми нарушениями в СА. Однако ясно, что UBЕ3A тесно связан с синаптической функцией нейронов.
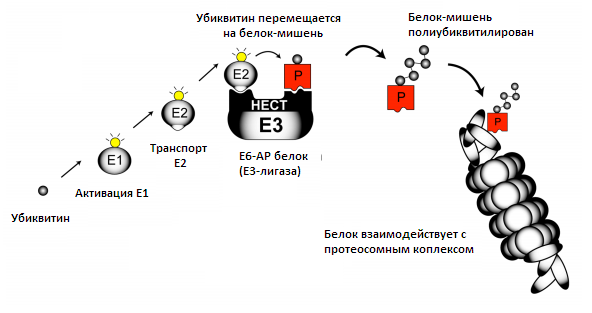 Рисунок 4. Путь образования убиквитин — протеосом
Рисунок 4. Путь образования убиквитин — протеосом
UBЕ3A и запечатление.
Известно, что UBE3A «запечатляется» в нейронах мозга [15, с. 837—47]. Это означает, что UBЕ3A ген из отцовской 15 хромосомы практически полностью не активен во многих областях мозга, а ген материнской 15 хромосомы обладает активностью. Нейроны мозга являются нормальными, несмотря на то, что у них есть только одна активная копия Гена UBЕ3A. То, что хромосомные делеции СА происходят только на материнской 15 хромосоме указывают на то, что UBE3A активен только на данной хромосоме, следовательно, делеция удаляет только активные копии гена. Нарушения генов, которые активны на отцовской 15 хромосоме становятся причиной развития другого расстройства, Синдрома Прадера-Вилли (ПВС). ПВС также предполагает запечатление генов, которые расположены близко, но отличаются от UBЕ3A. СА и ПВС являются довольно уникальными, потому что почти все другие генетические расстройства не проявляют этот тип запечатления
Термин «наследственное запечатление», возможно, труден для понимания. Для того, чтобы запечатленные гены нормально наследовались и работали в определенных хромосомах (например, как это происходит у здоровых людей), в них должен быть механизм для изменения экспрессии генов в определенное время развития яйцеклетки и эмбриона. Например, когда нормальный отец продуцирует сперму, независимо от того, отцовская или материнская 15 хромосома попадет в конечном итоге в сперматозоид, она должна быть «запечатлена» так, что ее гены становятся отключенными. Противоположные события происходят у здоровой матери, чьи яйцеклетки должны иметь все включенные гены. Запечатленные гены, таким образом, получают инструкции для активации стертыми.
Родословная показывает, как наследственное запечатление может стать причиной рецидива дальними родственниками: когда UBE3A мутация наследуется в семье, лица, которые наследуют мутации могут получить или не получить СА. Наследование UBЕ3A мутации от отца (верхний левый угол родословной) не имеет влияние на его детей, так как его UBЕ3A ген не активен. Не беда, если этот ген имеет мутации, поскольку каждый из его детей также унаследовал нормальную 15 хромосому от их матери. Однако если его дочь передает мутацию UBE3A своему ребенку, он будет болен СА, так как ребенок получит инактивированные UBE3A от ее отца.
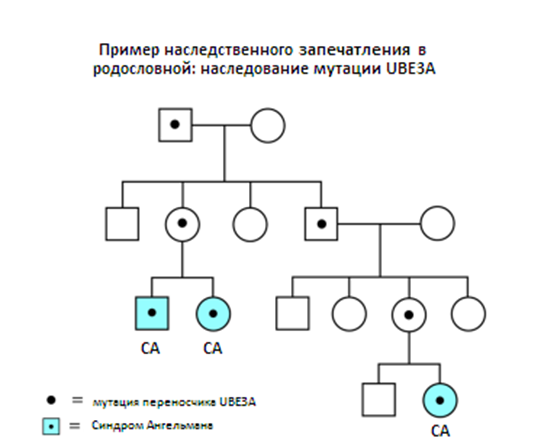
Рисунок 5. Пример наследственного запечатления в родословной
Генетические механизмы и интенсивность симптомов.
Все генетические механизмы СА приводят к достаточно единообразной клинической картине: тяжелая или глубокая умственная отсталость, характерное поведения, тяжелые нарушения речи, однако, имеются некоторые клинические различия, которые коррелируют с генотипом, несмотря на большую изменчивость в пределах каждой группы [1, с. 35—40; 4, с. 7—322; 5, с. 44—638; 8, с. 45—834; 11, с. 60—554; 12; 55—2547].
1. Делеция наиболее сильно связана с микроцефалией, судорогами, гипопигментацией, моторными нарушениями (например, атаксия, мышечная гипотония, трудности в кормлении), когнитивные и языковые нарущения;
2. ООД и ДЗ.Пациенты имеют неплохой физический рост (например, меньше шансов на микроцефалию) и имеют менее выраженные аномалии движения и атаксию (но не отсутствие);
3. ДЗ.Группа, как правило, имеет высокие когнитивные способности, восприятие речи, хорошую мелкую моторику и двигательные способности по сравнению с другими подтипами. Наиболее продвинутая речь наблюдается в этой группе (около 20 % группы) [12, с. 2547]. Эти люди могут говорить до 50—60 слов и использовать простые предложения;
4. Мутация UBE3A. Группа в целом занимает промежуточное положение между делецией и ДЗ по критериям: микроцефалия, судороги, двигательные нарушения, и речевые способности. Некоторые с мутацией UBЕ3A могут иметь относительно высокиекогнитивные способности, мелкую моторику, и грубые моторные навыки.
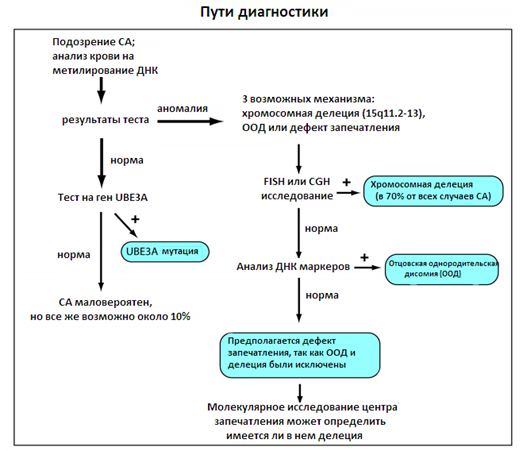
Рисунок 6. Пути диагностики синдрома Ангельмана
Лабораторное обследование на СА довольно сложное. Первый этап лабораторной оценки начинается с анализа на метилирование ДНК СА/ПВС региона. Если тест положительный, то присутствует один из трех механизмов: делеция, однородительская дисомия или дефект запечатления, и необходимы дополнительные исследования. В таких ситуациях следующий шаг обычно заключается в выполнении FISH, хромосомного теста, позволяющего увидеть, присутствуют ли делеции15q11.2-13. Есть другие методы для обнаружения этой делеции, например, на основе массива сравнительной геномной гибридизации. Если тест нормальный, то следующий шаг заключается в поиске отцовской однородительской дисомии путем дополнительных тестирований с привлечением родительских данных. Люди с позитивным тестом на метилирование ДНК, которые имеют нормальный FISH и нормальный тест на ООД, предположительно имеют дефект запечатления. Если тест на метилирование отрицательный, анализ мутации UBЕ3A может обнаружить нарушения.
Список литературы:
1. Bottani A., et al., Angelman syndrome due to paternal uniparental disomy of chromosome 15: a milder phenotype? Am J Med Genet, 1994.
2. Charles A. Williams; Sarika U. Peters; Stephen N. Calculator. http://www.angelman.org/_angelman/assets/File/.
3. Facts about Angelman syndrome. September 29, 2012.
4. Fridman C., et al., Paternal UPD15: further genetic and clinical studies in four Angelman syndrome patients. Am J Med Genet, 2000.
5. Gillessen-Kaesbach G., A previously unrecognised phenotype characterised by obesity, muscular hypotonia, and ability to speak in patients with Angelman syndrome caused by an imprinting defect. EurJ Hum Genet, 1999.
6. Jiang Y., Genetics of Angelman syndrome. AmJ Hum Genet, 1999.
7. Kishino Т., M. Lalande, and J. Wagstaff, UBЕ3A/Е6-AP mutations cause Angelman syndrome Nat Genet, 1997.
8. Lossie A.C., et al..Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet, 2001.
9. Mann M.R. and M.S. Bartolomei, Towards a molecular understanding of Prader-Willi and Angelman Syndromes. Hum Mol Genet, 1999.
10. Matsuura Т., et al., De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBC3A) in Angelman syndrome. Nat Genet, 1997.
11. Moncla A., Angelman syndrome resulting from UBE3A mutations in 14 patients from 8 families: clinical manifestations and genetic counselling. J Med Genet, 1999.
12. Nazlican H., et al. Somatic mosaicism in patients with Angelman syndrome and an imprinting defect. Hum Mol Genet, 2004.
13. Scheffner M., U. Nuber, and J.M. Huibregtse, Protein ubiquitination involing an E1-E2-E3 enzyme ubiquilinthioesler cascade. Nature, 1995.
14. Verdecia M.A., et al..Conformational flexibility underlies ubiquilin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell, 2003.
15. Yamasaki K., et al. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet, 2003.
