МОЛЕКУЛЯРНО-ДИНАМИЧЕСКОЕ МОДЕЛИРОВАНИЕ ОСАЖДЕНИЯ БИМЕТАЛЛИЧЕСКИХ КЛАСТЕРОВ НА ПОДЛОЖКУ
Конференция: LXXXIV Международная научно-практическая конференция «Научный форум: технические и физико-математические науки»
Секция: Физика конденсированного состояния

LXXXIV Международная научно-практическая конференция «Научный форум: технические и физико-математические науки»

МОЛЕКУЛЯРНО-ДИНАМИЧЕСКОЕ МОДЕЛИРОВАНИЕ ОСАЖДЕНИЯ БИМЕТАЛЛИЧЕСКИХ КЛАСТЕРОВ НА ПОДЛОЖКУ
MOLECULAR DYNAMICS SIMULATION OF DEPOSITION OF BIMETALLIC CLUSTERS ON A SUBSTRATE
Akbarali Rasulov
Doctor of Physico-Mathematical Sciences, Professor, Ferghana State Technical University, Republic of Uzbekistan, Ferghana
Nodirbek Ibrokhimov
Candidate of Physical and Mathematical Sciences, doctoral student of DSc, Ferghana State Technical University, Republic of Uzbekistan, Ferghana
Zhakhongir Khodzhimatov
PhD student, Ferghana State Technical University, Republic of Uzbekistan, Ferghana
Azamat Tukhtasinov
PhD student, Ferghana State Technical University, Republic of Uzbekistan, Ferghana
Аннотация. В статье разработаны алгоритмы и компьютерная программа, основанная на методы молекулярной динамики (МD) и метод Монте-Карло (МК), для описания процесса осаждения кластеров CoxAgх на поверхность Ag(100) при начальных энергиях (0.25-1.5 эВ/атом). Компьютерным моделированием построены равновесные конфигурации металлических кластеров CoxAgх при температуре T=0 K. Проведено осаждение малых и больших атомных кластеров на поверхность кристалла серебра. Результаты моделирования показывают, что проникновение кластеров в мишень зависит как от их энергии, так и от их размеров, причем зависимость от размеров сильнее. Более крупный кластер проникает на меньшую глубину, чем кластер меньших размеров. Меньший кластер практически полностью сливается с мишенью.
Abstract. The article develops algorithms and a computer program based on the methods of molecular dynamics (MD) and the Monte Carlo method (MC) to describe the deposition of clusters of CoxAgх on the surface of Ag(100) at initial energies (0.25-1.5 eV/atom). The equilibrium configurations of the metal clusters of the CoxAgх were constructed by computer modeling at a temperature of T=0 K. The deposition of small and large atomic clusters on the surface of a silver crystal is carried out. The simulation results show that the penetration of clusters into the target depends on both their energy and their size, and the size dependence is stronger. A larger cluster penetrates to a lower depth than a smaller cluster. The smaller cluster almost completely merges with the target.
Ключевые слова: биметаллические кластеры, наноструктура, моделирование, низкая энергия, термодинамическое равновесие.
Keywords: bimetallic clusters, nanostructures, modeling, low energy, thermodynamic equilibrium.
Введение
Наночастицы обладают уникальными свойствами, определяемыми их размером и квантово-механической природой. Особенно интересны биметаллические нанокластеры, сочетающие свойства двух различных элементов. Такие структуры перспективны в нанотехнологиях, оптике и магнитных устройствах.
Ранее проводились как экспериментальные, так и численные исследования структуры и стабильности кластеров Ag–Co [1, 2]. Особое внимание уделяется использованию классических моделей, которые позволяют моделировать системы с числом атомов в сотни тысяч [3].
Материалы и методы
Для моделирования использовалась молекулярная динамика с потенциалом EAM [4] и алгоритмами MPI. Учтено электрон-фононное взаимодействие, особенно значимое для охлаждения системы после удара [5]. В случае биметаллических кластеров Ag–Co взаимодействие моделировалось только для серебра.
Метод Монте-Карло по Метрополису применялся для получения равновесных конфигураций кластеров CoxAg201−x в диапазоне температур 100–1000 K [6].
Результаты и обсуждение
С использованием параллельного программирования и MPI кластеры ConAgm с n = m (где n = 100, 250, 500, 750, 1000, 1250 и 1500) были осаждены на поверхность Ag(100) при энергии 0,25 эВ до 1,5 эВ на атом для исследования процессов роста тонкой плёнки методом осаждения кластеров с низкой энергией (LECBD). В этом случае кластеры ConAgm с количеством атомов 200, 500, 1000, 1500, 2000, 2500 и 3000 осаждаются последовательно, случайным образом выбирая следующий кластер из указанного списка.
Результаты показали, что меньшие кластеры сильнее деформируются и глубже проникают в подложку, тогда как крупные кластеры сохраняют внутреннюю структуру [7]. На низких температурах атомы Co группируются под поверхностью, избегая центра, при x < 20. При x > 50 кобальт концентрируется в центре, но смешение с Ag остаётся ограниченным. Промежуточные стехиометрии демонстрируют либо группы Co под поверхностью, либо центральное ядро.
Кластеры размером до 3000 атомов использовались для анализа условий формирования структуры ядро–оболочка и фазовых переходов [8].
В результате конкуренции между напряжением и связыванием кобальта при низких температурах, при x<20 атомы кобальта располагаются непосредственно под поверхностным слоем кластера группами не более пяти атомов, занимая чётко определённые позиции и избегая центральной области кластера. Повышение температуры способствует укрупнению этих групп. Их растворение предсказывается при температурах, превышающих температуру плавления кластера. При x>50 атомы кобальта формируют скопление в центре кластера и пересекают грани {111}, когда серебряных атомов недостаточно для образования полной оболочки. При этих стехиометриях температура оказывается недостаточной для смешения Ag и Co даже выше температуры плавления. При осаждении маленьких кластеров (например, Co10Ag191) их форма нарушается, формируется интерфейс с ориентациями <100> и <110>. На высоких энергиях выравнивание затруднено из-за имплантации. Повреждение подложки варьируется в зависимости от энергии, образуются ступеньки и островки из атомов [9]. Учитывая морфологию и стехиометрию, можно контролировать поведение кластеров на атомном уровне.
|
|
|
|
Рисунок 1. Конфигурация кластера Co10Аg191 (маленький) и Co285Аg301 (большой). |
|

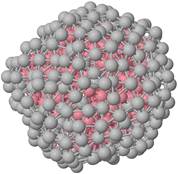
Кластеры аналогичного диапазона размеров используются для моделирования их замедления при осаждении на подложку. Замедление нанокластеров Co10Ag191 и Co285Ag301 на поверхности Ag(100) исследуется на атомарном уровне с использованием классического молекулярно-динамического моделирования.
Заключение
Использование метода MD с потенциалом EAM подтвердило свою эффективность для анализа осаждения кластеров Co-Ag. Выявлено, что:
- Меньшие кластеры глубже проникают и интегрируются в подложку;
- Для сохранения структуры требуется энергия ≤ 0.25 эВ/атом или крупные кластеры (Co > 250);
- Размер оказывает большее влияние, чем энергия.
Проведённые исследования способствуют пониманию морфологии и свойств наноструктурированных материалов, получаемых методом осаждения кластеров [10].
