Разработка компьютерных моделей для отбора наиболее перспективных потенциальных ингибиторов сборки вирусных частиц
Конференция: XVIII Международная научно-практическая конференция «Научный форум: медицина, биология и химия»
Секция: Технология получения лекарств

XVIII Международная научно-практическая конференция «Научный форум: медицина, биология и химия»
Разработка компьютерных моделей для отбора наиболее перспективных потенциальных ингибиторов сборки вирусных частиц
Development of the computer models for the most perspective potential viral particles assembly inhibitors selection
Vladimir Aladinskiy
Researcher, ChemRar Research and Development Institute, LLC, Russia, Khimki
Anastasia Aladinskaya
Researcher, ChemRar Research and Development Institute, LLC, Russia, Khimki
Mark Veselov
Researcher, ChemRar Research and Development Institute, LLC, Russia, Khimki
Andrey Ayginin
Junior Researcher, ChemRar Research and Development Institute, LLC, Russia, Khimki
Aleksei Riakhovskii
Ph.D. in Biological Sciences, Senior, Head of department, ChemRar Research and Development Institute, LLC, Russia, Khimki
Работа выполнена в рамках соглашения c «Минобрнауки России» о предоставлении субсидии от 26 сентября 2017 года №14.579.21.0154 (шифр заявки "2017-14-579-0057-130", уникальный идентификатор проекта RFMEFI57917X0154).
Аннотация. В последнее время с целью лечения гепатита B одной из стратегий стала разработка новых препаратов, связывающихся с капсидом вируса гепатита B (ВГВ) [1, 2]. Нами рассмотрены успехи и перспективы в данном направлении и разработаны фармакофорные модели для отбора потенциальных ингибиторов HBcAg методами виртуального скрининга для последующего подтверждения их действия in vitro.
Abstract. Recently one of the strategies with aim of hepatitis B treatment has became developing of new compounds that bind to the capsid of the hepatitis B (HBV) virus [1, 2]. We have described the progress and the perspectives on this way and We have developed pharmacophore models for the potential HBsAg inhibitors selection by virtual screening for further in vitro confirmation of their activity.
Ключевые слова: вирус гепатита В; ингибиторы HBcAg; фармакофорная модель.
Keywords: hepatitis B virus; HBcAg inhibitors; pharmacophore model.
Введение. Разработанная учеными в Bayer молекула BAY 41-4109 помимо активного ингибирования репликации вируса гепатита B продемонстрировала значительное снижение уровня кор-белка гепатита B (HBcAg) в печени трансгенных мышей, позволяя таким образом предположить совершенно новый механизм действия молекулы. Исследования показали, что BAY41-4109 вызывает неправильную сборку кор-белковых капсидов, таким образом, снижая их стабильность [3]. Однако дальнейшая разработка BAY 41-4109 была приостановлена по причине вызванной in vitro и in vivo токсичности и низкой эффективности в животных моделях [4, 5].
Двадцатигранная структура капсида вируса гепатита B состоит из одного вида белков – кор-белков гепатита Б, которые предположительно существуют в виде гомодимеров. Однако кор-белок всегда выделяется в виде димерной структуры. Капсид – стабильный полимер, состоящий из 90-120 гомодимеров кор-белка, способный образовываться самостоятельно в условиях in vitro.
Во время самосборки, гомодимеры кор-белка взаимодействуют друг с другом, по неизвестному согласованному механизму, вследствие которого образуются нуклеокапсиды, с заключенными внутри вирусной pgРНК и полимеразой гепатита B. Таким образом, внутри капсида происходит транскрипция и репликация, с образованием вирусной ДНК гепатита B [6]. Кор-антиген, также известный как HBcAg, преставляет собой полипептидную цепь, длинной в 183 аминокислотных остатка. Первые 149 аминокислот формируют формируют N‑терминальный домен (NTD), тогда как остальные 34 аминокислоты образуют C-терминальный домен (CTD) [7].
Гетероарилдигидропиримидины (ГАП), в список которых входит BAY41-4109, являются самой исследованной группой химических соединений, из класса молекул, способных вызывать нарушение сборки нуклеокапсида гепатита B (Рисунок 1). Среди активных аналогов ГАП можно выделить несколько схожих фрагментов, предположительно влияющих на активность данных соединений. Так, в позиции 4 обычно располагается галоген-замещенная фенильная группа, с предпочтительной R стерео конфигурацией. В позициях 2 и 5 предпочтительно расположение гетероцикла и сложного эфира соответственно. Позиция 6 – является важным участком для оптимизации молекулы, влияя на активность соединения in vitro и фармакокинетические свойства. К примеру, соединения GLS-4 и NVR-010-001, представляющие новое поколение аналогов ГАП, с 6 - морфолин заместителем, продемонстрировали увеличение in vitro активности в 5 раз по сравнению с BAY41-4109 [8, 9].
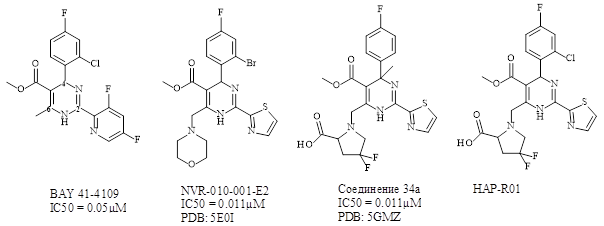
Рисунок 1. Соединения, с центральным ГАП-фрагментом. В BAY 41-4109 цифрами отмечены позиции в пиримидине
Одной из сложностей в оптимизации ГАП соединений являлось отсутствие кристаллических структур высокого разрешения. Так капсиды вируса гепатита B были получены в разрешении 3Å, тогда как структуры включающие малые молекулы – 4.2 и 5.05 Å соответственно. Недавно были получены новые рентгеноструктурные данные в высоком разрешении диапазоном 1.5-2Å. Для построения наиболее точной модели, нами были проанализированы все кристаллические структуры, представленные в Таблице 1.
Таблица 1.
Рентгеноструктурные данные кор-белков в высоком разрешении
|
Номер PDB |
Разрешение |
Лиганд |
Дата публикации (мес.год) |
|
5E0I |
1.95Å |
NVR 10-001E2 |
12.2015 |
|
5GMZ |
1.7Å |
34а |
08.2016 |
|
5WRE |
1.95Å |
HAP-R01 |
02.2017 |
|
5T2P |
1.69Å |
SBA-R01 |
02.2017 |
Изучение кристаллической структуры содержащей ингибитор NVR 10-001E2 (5E0I). Полученные в 2015 году, первые рентгеноструктурные данные связывания кор-белка с ГАП-лигандом в высоком разрешении, предоставили новые возможности в разработке и оптимизации лигандов кор-белка [10]. Было показано, что наличие ГАП-ингибитора NVR-010-001-E2 в структуре Y132A мутированного белка (CoreND-Y132A) позволяет увеличить качество разрешения до 1.95 Å, в отличие от 3Å в его отсутствие. Тирозин в позиции 132 является ключевой аминокислотой, определяющей стабильность взаимодействия между кор-белковыми димерами гепатита B и образование икосаэдрического капсида. Так, мутация, посредством которой происходит замещение тирозина на аланин, приводит к неспособности белков образовывать капсиды, при этом сохраняя возможность взаимодействия с кор-белками [11, 12]. В кристаллической структуре (PDB номер:5E0I) представлены три сайта димер-димерных взаимодействий и три сайта гексамер-гексамерных взаимодействий. Наибольший интерес представляет область взаимодействия в димер-димерном участке, где лиганд находится внутри кармана связывания, о котором далее пойдем речь. Внутри сайта связывания расположен гидрофобный карман, образованный одной белковой субъединицей, тогда как C-концевые альфа спираль (α5) и петля соседней белковой субъединицы образуют дополнительную область для взаимодействия с молекулой.
Центральный пиримидин в соединении NVR-010-001-E2 находится на нижней части поверхности связывания и «прикрепляется» к белку посредством водородной связи между одним из азотов и боковой цепью триптофана 102 (W102). Бромфтор-замещенная фенильная группа простирается в гидрофобный карман, образованный пролином 25 (P25), гидрофобным участком аспаргиновой кислоты (D29), лейцином 30 (L30), и треонином 33 (T33) из альфа-спирали 4 (α4), а также триптофаном 102 (W102), изолейцином 105 (I105), и серином 105 (S105) из альфа-спирали 2 (α2). При этом бром направлен глубоко внутрь гидрофобного кармана. Фтор-заместитель на фенильном кольце взаимодействует с прилежащей поверхностью, а именно валином 124 (V124’), алкильной цепью аргинина 127 (R127’) и треонином 128 (T128’) (аминокислоты, входящие в состав прилежащей поверхности обозначаются с апострофом). Тиазол располагается внутри гидрофобного участка, образованного ароматическими группами триптофана 102 (W102), фенилаланина 122 (F122) и тирозина 118 (Y118). Азот, расположенный в тиазольном гетероцикле, образует водородную связь с боковой цепью лейцина 140 (L140), опосредованную молекулой воды. Также наблюдаются взаимодействия между тиазолом и треонином 128 (T128’), а также эфирной функциональной группой и валином 124 (V124’). Основные взаимодействия морфолина возникают с поверхностью соседнего димера, образованного петлей и α5 спиралью, а именно с аминокислотными остатками V124’, W125’, W128’ и P134’. Полученная кристаллическая структура позволила предположить, что улучшение противовирусной (ВГВ) активности NVR 010-001-E2 по сравнению с BAY 41-4109 может быть связано с дополнительными взаимодействиями морфолинового заместителя с поверхностью другого димера кор-белка, соответственно повышая стабильность димер-димерных взаимодействий, и соответственно инициировать образование стабильных белковых структур.
Изучение кристаллической структуры, содержащей 4-метил-ГАП-структурный лиганд (5GWZ). Структурно-функциональный анализ структур с 4-метил-ГАП фрагментом был недавно представлен исследовательской группой фармацевтической компании Roche [13]. Указанная серия соединений была разработана с целью оптимизации BAY41-4109. Несмотря на то, что большинство наномолярных ингибиторов сборки капсида с ГАП центральным фрагментом содержат в 4 положении пиримидина -фенил с заместителями во 2 и 4 положениях, 2-хлор и 2-фтор замещение в том же положении в серии 4-метил ГАП соединений приводит к снижению активности и повышению цитотоксичности. Однако наличие пара-заместителя в фениле в 4 положении, является необходимым условием для получения противовирусной активности соединения. Стоит отметить, что замена фтора на –H, -SO2Me, -OMe и –CN приводит к снижению активности in vitro.
При этом, как и ожидалось, наличие полярного электроноакцепторного заместителя в пара-положении, приводит к улучшению метаболической стабильности и растворимости молекулы. При дальнейшей оптимизации в 6 положении, было показано что морфолин, позволяет значительно увеличить активность. Так, наибольшая активность при оптимизации в 6 положении наблюдалась для 3,4 фтор –замещенного метил-аналога ГАП (Рисунок 2).
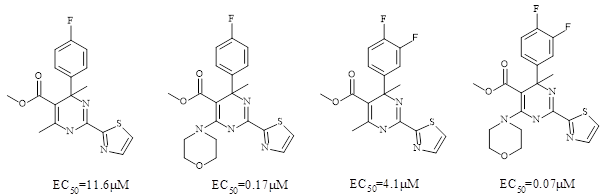
Рисунок 2. Структуры соединений из серии 4-метил-ГАП. Оптимизация в положении 6. EC50 указана для (S)-энантиомеров
Стандартные «биоизостерные» замены морфолина, такие как пиперидин и пиперазин приводили к полной потере активности соединений. Однако при замене такими функциональными группами как гем-диметил морфолин и 3,3-дифторпирролидин наблюдается наномолярная активность соединений (Рисунок 3).
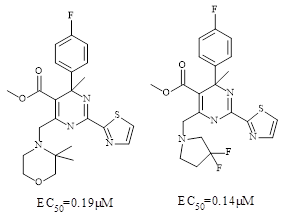
Рисунок 3. Структуры соединений из серии 4-метил-ГАП. Оптимизация в положении 6
Недостатком этих функциональных групп является их липофильный характер, что снижает растворимость молекулы, соответственно снижая потенциал и перспективность дальнейших исследований in vivo. Добавление карбонильной группы в структуру указанных фрагментов привело к получению соединений с высоким показателем активности. После указанных стадий оптимизации было найдено наиболее перспективное соединение 34a (Рисунок 1). Интересно, что показатель селективности полученного соединения был значительно лучше, чем для BAY 41-4109. Кристаллическая структура комплекса Y132A мутантного белкового капсида с соединением 34а была получена в высоком разрешении (1.7 Å) (PDB номер: 5GMZ). Из представленной структуры видно, что тиазол лежит в одной плоскости с дигидропиримидиновым кор-фрагментом, располагаясь в гидрофобном кармане, образованном фенилаланином 23 (F23), P25, Y118, T128' и аланином 132 (A132'). Стоит отметить, что 4-метил и пара-фторфенил находятся в смежном гидрофобном кармане, и образовывают Ван-дер-Ваальсовы взаимодействия с аминокислотами P25, L30, T33, W102, I105, серином 106 (S106), V124', R127' T128'. Из кристаллической структуры видно, что пространство вокруг фторфенильной группы ограничено, объясняя описанное ранее снижение активности соединений с дополнительными 2‑хлор и 2 фтор заместителями. Так же как и в кристалле 5E0I была отмечена водородная связь между азотом пиримидинового структурного фрагмента и W102. Важно отметить, что метильная группа эфира в 5 положении взаимодействует с наружной областью кармана, образованной лейцином 37 (L37) и треонином 109 (T109). Таким образом, наличие больших полярных групп в данном положении нежелательно. Фтор заместители, входящие в состав 4,4 – дифторпролиновой функциональной группы, взаимодействуют с W125’, T128’, аргинином 133 (R133’) и P134’. Также между карбоксильной кислотой и серином 141 (S141’) образовывается бинедатная водородная связь. Возможно, что взаимодействия функциональной группы в положении 6 оказывают значительное влияние на связывание соединения с белковым капсидом, способствуя, таким образом, повышению активности против ВГВ. Недавно была опубликована кристаллическая структура, отражающая механизм связывания ГАП-содержащего ингибитора (HBA-RO1), модифицированного в 6 положении при помощи функциональной группы, идентичной соединению 34а (PDB номер:5GMZ). Более того, была получена кристаллическая структура комплекса Y132A мутантного белкового капсида с лигандом SBA-R01 (Рисунок 4).
Изучение кристаллической структуры, содержащей сульфамоил-бензамид-структурный лиганд SBA-R01 (5T2P). Соединение SBA-R01 относится к типу сульфамоил-бензамидов (СМБ), которые представляют совершенно другой класс модуляторов образования капсида. Молекулы, входящие в класс соединений ГАП являются ингибиторами сборки вирусных капсидов, тогда как СМБ препятствуют инкапсуляции вирусной РНК, таким образом, приводя к образованию "пустых" капсидов. Стоит отметить, так же, как и в случае HBA-RO1, SBA-R01 образовывает водородную связь с W102, при этом 3, 4, 5-трифторфенил занимает тот же гидрофобный карман, что и галоген-замещенная фенильная группа в случае HBA-R01. Однако SB-R01 не занимает дополнительный гидрофобный карман в отличие от HBA-RO1. Также, центральные фенильные структурные фрагменты не накладываются друг на друга, в случае наложения кристаллов, находясь в разных плоскостях.
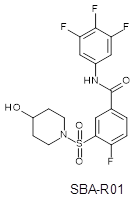
Рисунок 4. Структура СМБ - структурного ингибитора SBA-R01
Разработка фармакофорной модели для поиска потенциальных ингибиторов HBcAg. Для построения модели были использованы кристаллические структуры под номерами 5E0I, 5GMZ, 5WRE, 52TP полученные из базы данных PDB (Protein Data Bank) [14]. Представленные белки были препроцессированы при помощи «Protein Preparation Wizard» в программе Schrödinger Maestro [15]. После чего, было осуществлено наложение структур при помощи «Protein Structure Alignment» (Рисунок 5).
Построение фармакофорной гипотезы было осуществлено в программе Molecular Operating Environment (MOE) [16]. На основании анализа кристаллических структур было построено 5 гипотез. Последующая оптимизация позволила остановиться на трех, наиболее оптимальных моделях позволяющих отобрать молекулы с необходимой геометрией и структурой. В каждой модели были учтены описанные выше результаты теоретического исследования и экспертная оценка кристаллических структур.
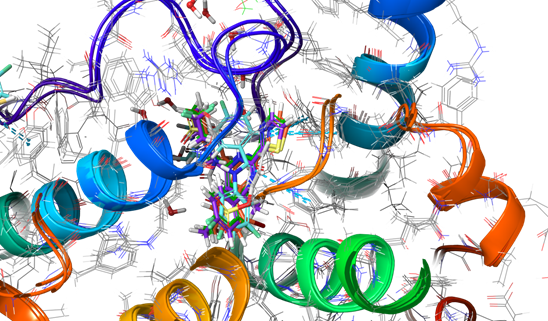
Рисунок 5. Наложение кристаллических структур 5E0I, 5GMZ, 5WRE, 52TP
При построении первой гипотезы, акцент был сделан на гидрофобной области в кармане связывания, взаимодействие с которой играет ключевую роль в связывании. Так во всех разработанных на данный момент модуляторах сборки капсида присутствует фрагмент, располагающийся в данной области сайта связывания. Целью было найти соединения с присущей геометрией в положении 4. Акцепторный фрагмент также был включен в гипотезу с целью поиска соединений потенциально способных к образованию водородной связи с L140. Для построения, было использовано наложение всех кристаллических структур, представленных в Таблице 1. Для фармакофорной гипотезы №1 (Рисунок 6) были выбраны: (1) ароматическая группа в 4 положении, (2) гидрофобный участок в 4 положении, (3) пара заместитель фенила, расположенного в 4 положении, гидрофобный атом, (4) акцептор, входящий в состав центрального пиримидина, (5) гидрофобный участок в 6 положении, (6) исключенный объем, обозначающий центр связывания и воду в кармане связывания, опосредующую образованию водородных связей между активной молекулой и белком. На всех соответствующих рисунках сферами представлены центры фармакофорной гипотезы: оранжевый – ароматический фрагмент, зеленый – гидрофобная область, желтый – гидрофобный атом, голубой – наличие акцептора в заданной области.
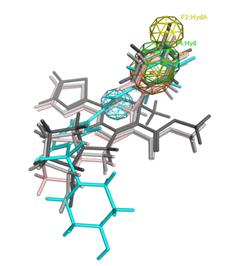
Рисунок 6. Фармакофорная гипотеза № 1
При построении фармакофорной гипотезы №2, была учтена также гидрофобная область молекулы на выходе из кармана. Для фармакофорной гипотезы 2 (Рисунок 7) были выбраны: (1) ароматическая группа в 4 положении, (2) гидрофобный участок в 4 положении, (3) пара заместитель фенила, расположенного в 4 положении, гидрофобный участок, (4) акцептор, входящий в состав центрального пиримидина, (5) гидрофобный участок в 6 положении, (6) исключенный объем, обозначающий центр связывания и воду в кармане связывания, опосредующую образованию водородных связей между активной молекулой и белком.
При построении фармакофорной гипотезы 3 решено было обратить внимание исключительно на ГАП-структурные ингибиторы, исключив из наложения кристалл, содержащий СМБ структурный ингибитор (Рисунок 8). Несмотря на то, что SBA-01 взаимодействует с тем же центром связывания, механизм воздействия, как указано выше для данной группы соединений другой.
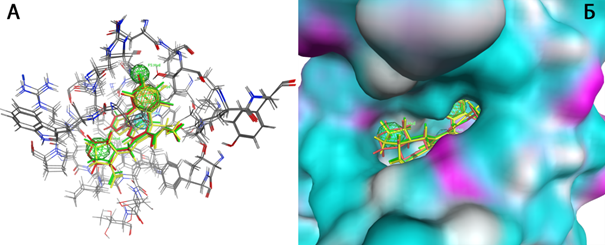
Рисунок 7. А) Фармакофорная гипотеза № 2 Исключенные объемы не представлены с целью улучшить информативность изображения. Б) Представление фармакофорной гипотезы в кармане связывания. Голубым цветом отмечена гидрофобная область поверхности. Ингибиторы сборки капсида представлены в разных цветах
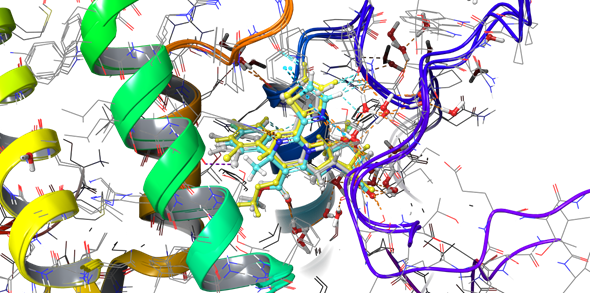
Рисунок 8. Наложение кристаллических структур 5E0I, 5GMZ, 5WRE, включающих в свою структуру ГАП ингибиторы
В качестве фармакофорных центров гипотезы № 3 (Рисунок 9) были выбраны: (1) ароматическая группа в 4 положении, (2) пара заместитель фенила, расположенного в 4 положении, (3) акцептор, входящий в состав центрального пиримидина, (4) акцептор в составе тиазола (5) гидрофобный участок в 4 положении, (6) исключенный объем структуры кармана и консервативной воды в центре связывания.
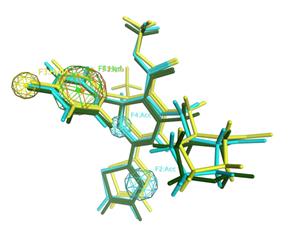
Рисунок 9. Фармакофорная гипотеза №3
Заключение. В ходе теоретического исследования были изучены структурные особенности представленных мишеней, а именно центров связывания. В работе были изучены все научные публикации по данным тематикам представленные за последние 5 лет. Также, были исследованы структуры и особенности взаимодействия известных ингибиторов сборки капсида и ингибиторов входа ВГВ.
Появление структур высокого разрешения стало ключевым фактором в определении взаимодействий и оптимизации потенциальных ингибиторов сборки капсида с мишенью. Представленный в 2015 метод точечной мутации Y132A лег в основу всех рентгеноструктурных данных высокого разрешения, представленных на текущий момент.
ГАП являются наиболее известной и изученной группой соединений из представленных ингибиторов сборки капсида. Практически все полученные кристаллы содержат в своей структуре ГАП-структурные ингибиторы. Механизм действия данной группы молекул основан на том, что связывание ингибиторов с областью взаимодействия кор-белков приводит к неправильной сборке капсида, нарушая, таким образом, его функциональность. Недавно впервые были получены рентгеноструктурные данные для СМБ-структурного ингибитора (SBA-R01). В отличие от ГАП, СМБ приводит к «правильной» сборке капсида который, однако, является «пустым» и не содержит в своей структуре вирусной РНК. Примечательно, что полученные данные продемонстрировали, что SBA-R01 взаимодействует с тем же карманом связывания, что и ГАП. Таким образом, при построении первой и второй фармакофорных гипотез, были использованы наложения всех четырех кристаллических структур, включая СМБ-содержащую. На основании полученного наложения, были отмечены фармакофорные центры на мотивах ингибиторов, отмеченные как ключевые в механизме связывания. Так, акцент был сделан на гидрофобном кармане внутри центра связывания. Однако далее решено было также построить дополнительную гипотезу, с целью выделить, дополнительно группу соединений, действующих предположительно по механизму ГАП. Полученная гипотеза отличалась, наличием дополнительного акцепторного центра. Так, ранее было отмечено, что отличие в механизме ГАП, может быть связано с взаимодействием в дополнительном гидрофобном кармане центра связывания. В результате, были разработаны 3 фармакофорных гипотезы, направленных на поиск ингибиторов сборки капсида по двум ключевым механизмам, основанные на последних рентгеноструктурных данных высокого разрешения.
В отличие кор-белковых капсидов, кристаллографическая структура NTCP не получена на данный момент. С целью, повысить точность отбора соединений, решено было построить гомологичную модель, основанную на структуре ASBT (Номер PDB: 3ZUY). Далее была построена и проанализирована карта Рамачандрана трехмерной структуры. Так, подавляющее количество аминокислотных участков было расположено в минимуме энергии, что свидетельствует об умеренном качестве модели. Полученная гомологичная структура белка не позволяет прогнозировать активность ингибиторов с высокой точностью, однако может быть использована для понимания и определения ключевых взаимодействий. Полученная трехмерная модель позволила выделить 2 гидрофобных кармана, которые, исходя из структуры природных лигандов NTCP являются одними из ключевых участков в центре связывания. Фармакофорные гипотезы были построены на основе 18 известных ингибиторов входа для определения основных фармакофорных центров, а также 10 дополнительных для определения области исключенного объема. Были определены три фармакофорных гипотезы, которые в дальнейшем использовались для поиска виртуальных хитов, ингибиторов NTCP.
