СИНДРОМ АНГЕЛЬМАНА: ЭТИОЛОГИЯ, ПАТОГЕНЕЗ, КЛИНИКА, ДИАГНОСТИКА
Журнал: Научный журнал «Студенческий форум» выпуск №4(183)
Рубрика: Медицина и фармацевтика

Научный журнал «Студенческий форум» выпуск №4(183)
СИНДРОМ АНГЕЛЬМАНА: ЭТИОЛОГИЯ, ПАТОГЕНЕЗ, КЛИНИКА, ДИАГНОСТИКА
Аннотация. В данной статье раскрываются основные моменты этиологии, патогенеза и диагностики врожденной генетической аномалии - синдрома Ангельмана, обусловленного хромосомными нарушениями. Их существует несколько вариантов и все они связаны с хромосомным сегментом 15q11–q13. Основными проявлениями данного синдрома являются задержка физического развития, тяжелая умственная отсталость, немотивированный смех и проблемы с речью.
Ключевые слова: синдром Ангельмана, генетический синдром, хромосомы 15q11–q13.
Многие заболевания, причиной которых является генетическая мутация, проявляют себя уже в раннем детстве. Нарушения могут выявить сразу после рождения, или еще в пренатальном периоде, но некоторые признаки заболеваний возможно заметить только во время постнатального развития. К таким, например, относится синдром Ангельмана.
Синдром Ангельмана - наследственное заболевание, характеризующееся задержкой развития, умственной отсталостью, тяжелой задержкой речевого развития и атаксией [2]. Данное заболевание имеет много синонимов: его называют “синдромом Петрушки”, “синдромом счастливой марионетки” - это название дал британский педиатр Гарри Ангельман, который впервые сопоставил симптомы разных “детей-кукол” и описал их заболевание. Пациентов с этим синдромом очень ласково называют «дети ангелы» - за счастливую улыбку и особенности внешности.
Дети с синдромом Ангельмана рождаются с частотой один ребенок на 12-20 тысяч человек. Но нужно уточнить, что для постановки диагноза необходимо проведение молекулярно-цитогенетических исследований, поэтому бывают случаи, когда синдром не выявляется, а дети наблюдаются у невролога с задержкой речевого развития, нарушением поведения и эпилепсией [1].
Этиология и патогенез
Синдром Ангельмана является генетическим заболеванием, связанным с хромосомными аномалиями. Существует несколько вариантов нарушений, которые приводят к появлению синдрома. Наиболее часто встречающийся дефект - впервые возникшая делеция в локусе 15q11-q13. С делецией рождаются по разным источникам от 70% до 80% детей с синдромом Ангельмана. Вторым вариантом нарушений является отцовская дисомия по хромосоме 15, но встречается она гораздо реже - в 3-7% случаев. Следующий возможный вариант - это дефект центра импринтинга, частота встречаемости которого менее 3%. В 10% случаев синдрома Ангельмана нарушением являются мутации гена убиквитинлигазы UBE3A. Также у некоторых детей не удается идентифицировать молекулярную аномалию.
Таблица 1.
Частота встречаемости различных генетических дефектов у больных с синдромом Ангельмана [2]
|
Класс |
Генетический дефект |
Частота встречаемости |
|
Ia |
De novo делеция 15q11-q13 региона копии материнской хромосомы |
70-75% |
|
Ib |
Несбалансированная транслокация хромосомы |
< 1% |
|
IIa |
Отцовская дисомия по хромосоме 15 |
3-7% |
|
IIb |
Отцовская дисомия по хромосоме 15 с транслокацией |
< 1% |
|
IIIa |
Делеция импринтингового центра |
0,5% |
|
IIIb |
Нарушение импринтинга - эпимутация |
2,5% процентов пациентов с дефектом импринтинга |
|
IV |
Мутации UBE3A-гена |
≈ 10% |
|
V |
Молекулярные аномалии не идентифицируются |
10-15% |
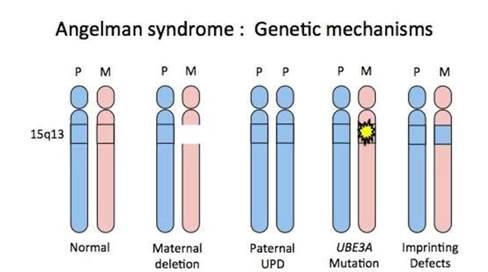
Рисунок. Генетические нарушения при синдроме Ангельмана
Морфологические или функциональные нарушения локуса q11-q13 копии материнской хромосомы 15 обусловливают потерю экспрессии гена UBE3A в нейронах головного мозга. Дефицит протеина UBE3A приводит к избыточному содержанию его целевых протеинов. Избыток протеинов ARC, эфексина-5 обусловливает нарушения цитоскелета дендритных шипов, что снижает синаптическую пластичность и проявляется когнитивными и поведенческими расстройствами, REST-ассоциированная дисфункция рецепторов гамма-аминомасляной кислоты обусловливает повышенную судорожную готовность, а избыточное содержание протеина BMAL1 приводит к нарушению сна и трофики [2].
Клиническая картина
Часто дети с генетическими заболеваниями имеют общие черты внешности. Так и дети с синдромом Ангельмана похожи. У большинства из них маленький череп, гипоплазия средней части лица, подбородок заострен, нижняя челюсть выступает вперед, большие промежутки между зубами и широкий рот, а глаза посажены глубоко, есть косоглазие. У ребенка могут быть “ангельские” черты лица - голубые глаза, волосы светлых оттенков, бледная кожа. Такие признаки могут появиться несмотря на то, что у родителей другой фенотип, это признак делеции в хромосоме.
Синдром Ангельмана проявляется уже в первые полгода жизни, но окончательно ставят диагноз гораздо позже. Как правило, родители таких детей впервые обращают внимание на задержку развития, например, на малый прирост окружности головы. Кроме этого, ребенок позже приобретает необходимые навыки, такие как держание головки, сидение. Чем старше он становится, тем более заметны отклонения. Появляются подергивания конечностей, резкие, неуклюжие движения, судороги. С развитием хождения становится заметна атаксическая походка с гиперкинезами, которые помогают поддерживать тело. Также появляются эпилептические припадки, пик которых приходится на детский возраст.
Речь у детей с синдромом Ангельмана начинает появляться очень поздно, но даже в старшем возрасте словарный запас не превышает 5-6 простых слов. Родители отмечают, что дети могут понимать обращенную к ним речь и различают эмоциональное состояние собеседника. При невозможности ответить словами дети учатся общаться жестами, у них хорошо развита способность к невербальной коммуникации. Обращает на себя внимание всегда счастливое выражение лица ребенка и частый необоснованный смех. Это является признаком поражения ствола мозга и не всегда отражает эмоции ребенка.
Отдельно выделяется еще одна особенность этих детей - малая потребность во сне. За ночь ребенок может спать по 4-5 часов, реже до 6 часов. Чаще всего сон прерывистый, с пробуждением и беспокойством. Днем дети чувствуют сонливость. Родителям тяжелее всего дается жить по такому графику с детьми, поэтому нарушения сна являются одной из самых главных жалоб.
Диагностика
Диагностика синдрома Ангельмана обычно проходит в несколько этапов. Из-за сложности этот процесс часто затягивается на несколько лет, а в некоторых случаях врачи в диагностике так и не доходят до истинной причины нарушений.
Первым с ребенком встречается педиатр, который при обнаружении различных неврологических нарушений направляет пациента на консультацию к неврологу. Этот специалист, в свою очередь, проводит обследование для поиска причины заболевания. Есть множество генетических заболеваний, которые проявляются похожими симптомами, поэтому так важна и необходима дифференциальная диагностика. Далее с ребенком знакомится врач-генетик, который и должен провести все нужные обследования и поставить окончательный диагноз. В ходе исследования ребенку назначают электроэнцефалографию, магнитно-резонансную томографию, а также молекулярно-цитогенетические исследования.
Научно-консультативный комитет фонда синдрома Ангельмана (Angelman Syndrome Foundation) в 2005 году выделил клинические критерии для диагностики синдрома. Все признаки были разделены на четыре группы по частоте встречаемости у больных. Первая группа - симптомы, полезные в качестве поддерживающих критериев, но отсутствие их не исключает диагноз. Вторая группа - обязательные признаки, которые встречаются у 100% больных (см. таб. №2). Третья и четвертая группы - соответственно часто встречающиеся признаки (присутствуют у более 80% больных) и дополнительные признаки (их имеют от 20 до 80% больных) [3].
Таблица 2.
Некоторые клинические критерии для диагностики синдрома Ангельмана [3]
|
Обязательные признаки (встречаются у 100 % пациентов с синдромом Ангельмана)
|
|
|
Часто встречающиеся признаки (встречаются более чем у 80 % пациентов с синдромом Ангельмана) |
|
Таким образом, своевременная генетическая диагностика для пациентов с синдромом Ангельмана имеет очень большое значение для их будущей жизни. Вовремя найденная патология дает больше шансов для успешной адаптации ребенка в обществе: детей с таким синдромом обучают языку жестов, также занимаются коррекцией их сна и режима, что особенно важно для всей семьи в целом, с ними занимаются врачи-ЛФК, логопеды и другие специалисты. Существенный вклад в жизнь семей с такими детьми приносят различные благотворительные организации, например, российский благотворительный фонд “Синдром Ангела”, который не только занимается финансовыми вопросами, но и оказывают моральную и психологическую помощь. Кроме этого, поддерживает научные исследования по поиску лекарства для детей с синдромом Ангельмана.
