Статья:




Клинический случай: Синдром Ангельмана
Секция: Медицина и фармацевтика
Выходные данные
Клименко А.Д. Клинический случай: Синдром Ангельмана // Естественные и медицинские науки. Студенческий научный форум: электр. сб. ст. по мат. V междунар. студ. науч.-практ. конф. № 5(5). URL: https://nauchforum.ru/archive/SNF_nature/5(5).pdf (дата обращения: 27.07.2026)
Лауреаты определены. Конференция завершена
Эта статья набрала 0 голосов
Мне нравится0
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников

V Студенческая международная научно-практическая конференция «Естественные и медицинские науки. Студенческий научный форум»
Клинический случай: Синдром Ангельмана
Клименко Александр Дмитриевич
студент, Ставропольский государственный медицинский университет, РФ, г. Ставрополь
Кадимова Зарина Малашерифовна
научный руководитель, врач- ординатор,
Ставропольский государственный медицинский университет,
РФ, г. Ставрополь
Минаева Ольга Александровна
научный руководитель, канд. мед. наук, доцент,
Ставропольский государственный медицинский университет,
РФ, г. Ставрополь
Колесникова Евгения Викторовна
научный руководитель, ассистент,
Ставропольский государственный медицинский университет,
РФ, г. Ставрополь
Среди клинически значимых хромосомных болезней у человека синдромы микроделеций имеют немаловажное значение.
По данным ВОЗ эта группа хромосомных аномалий вносит значимый вклад в структуру умственной отсталости. Микроделеционные синдромы (МДС) - это особый вид хромосомных заболеваний, при котором происходит потеря микроскопического участка хромосомного материала, что не удается зафиксировать рутинными методами цитогенетической диагностики.
К наиболее часто встречающимся МДС относятся CATCH-синдром, синдром микроделеции 1p36, синдром Прадера-Вилли, синдром Ангельмана, синдром Вольфа-Хиршхорна и другие. [1]
Синдром Ангельмана — это нейрогенетическое заболевание, характеризующееся задержкой интеллектуального и физического развития, нарушениями сна, приступами судорог, резкими движениями (особенно рук), частым беспричинным смехом или улыбкой и, как правило, эти люди, выглядят очень счастливыми.
Частота встречаемости данной патологии достаточно низкая и составляет 1 на 10000-20000 живорожденных младенцев. [3] Данный синдром имеет несколько названий: синдром смеющейся куклы, синдром счастливой куклы или синдром Петрушки, и все эти названия имеют право на существование, поскольку это связано с историей их описания.

Рисунок. 1 Доктор Г. Ангельман со своей женой
В 1965 году доктор Гарри Ангельман, английский врач, впервые описал трех детей с характеристиками, которые теперь известны как синдром Ангельмана (AS). Он отметил, что у всех была жесткая, вялая походка, речь отсутствовала, чрезмерный смех и судороги. Другие случаи в конечном итоге были опубликованы, но в то время, это состояние считалось крайне редким, и многие врачи сомневались в его существовании. Первые доклады из Северной Америки появились в начале 1980-х годов. Доктор Ангелман рассказывает следующее об открытии этого синдрома:
«История медицины полна интересных историй об открытии болезней. Сага о Синдроме Ангельмана - одна из таких историй. Было совершенно случайно, что почти тридцать лет назад (например, около 1964 года) трое детей-инвалидов неоднократно проходили лечение в моем отделении в Англии.
У них были различные нарушения, и хотя на первый взгляд они, казалось, страдали от различных состояний, но я чувствовал, что существует общая причина их болезни.
Диагноз был чисто клиническим, потому что, несмотря на технические исследования, которые на сегодня были наиболее совершенны, я не смог установить научное доказательство того, что трое детей имели одинаковые недостатки.
В связи с этим я не решался писать о них в медицинских журналах. Однако, когда на отдыхе в Италии мне довелось увидеть масляную живопись в музее Кастельвекьо в Вероне… «Мальчик с кукольником». Смеющееся лицо мальчика и то, что мои пациенты демонстрировали отрывистые движения, дали мне идею написать статью о трех детях с названием «Кукольные дети». Это было не имя, которое нравилось всем родителям, но оно служило средством объединения трех маленьких пациентов в одну группу. Позже название было изменено на синдром Ангельмана. Эта статья была опубликована в 1965 году и после этого первоначальные интересы были почти забыты до начала восьмидесятых годов. В 1987 году Эллен Магенис, врач Центра медицинских наук штата Орегон, выявила детей с микроделециями 15 хромосомы, которые, как ожидалось, имели синдром Прадера-Вилли. Тем не менее, у этих детей были изъяны и серьезная задержка развития, которые, как ожидалось, не будут найдены для этого синдрома. Было быстро осознано, что у этих детей были микроделеции на материнском числе 15, тогда как в синдроме Прадера-Вилли делеция всегда наблюдалось на родительском. Это было важным открытием и в конечном итоге проложило путь для разграничения нескольких механизмов, вызвавших AS, все из-за нарушения гена, расположенного на 15 хромосоме. Было выяснено, что синдром может быть вызван двумя копиями хромосомы по отцовской линии 15 (1991) и что регулирующий регион (Центр импринтинга) также могут быть нарушены до синдрома (1993). В 1997 году, спустя 10 лет после идентификации хромосомы, был выделен ген AS, UBE3A. Это открытие быстро привело к разработке моделей на животных и к активным исследованиям в области нейробиологии, направленным на выявление нарушений аномалий UBE3A в развитии нервной системы.
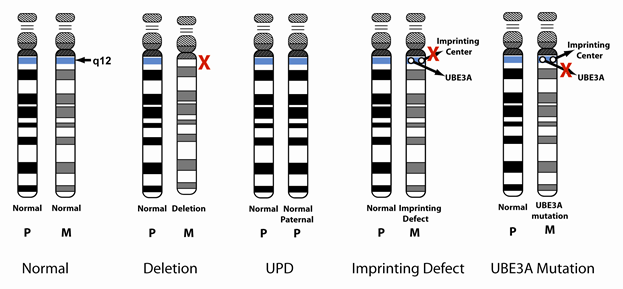
Рисунок. 2 Варианты повреждений
На сегодняшний день существуют несколько генетических механизмов синдрома, и все они связаны с хромосомным сегментом 15q11–q13: интерстициальные делеции 15-й хромосомы, доставшейся от матери; отцовская однородительская дисомия по 15-й хромосоме; мутация гена UBE3A, кодирующего убиквитин-протеин-лигазу. В остальных случаях точная причина болезни остается неизвестной. [4,5,6,7,9]
Клинические проявления
Для синдрома Ангельмана характерны:
• задержка психомоторного развития: тяжелая умственная отсталость с выраженной задержкой двигательного развития (100%), приступы немотивированного смеха, отсутствие речи или крайне малый набор слов — не более 6 (100%);
• фенотип: микробрахицефалия, светлые волосы (65%), глубоко посаженные глаза; аномалии глаз (в том числе гипопигментация сосудистой оболочки и радужки — у 88% больных светло-голубые глаза); косоглазие (42%); верхняя микрогнотия; большой рот с высунутым языком и широкими межзубными промежутками; нижняя прогнатия;
• со стороны центральной нервной системы: атаксия, частые хаотичные движения руками, подергивание рук при ходьбе, напоминающие движения марионетки (100%); эпилептические припадки, от больших до акинетических, чаще начинающиеся в 18–24 мес (86%); мышечная гипотония; иногда гиперрефлексия, леворукость.
Синдром Ангельмана является врожденной генетической аномалией; в настоящее время специфические способы его лечения не разработаны. Однако, некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии). Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных, эпилептические приступы — приемом противосудорожных препаратов; нарушения стула регулируются назначением легких слабительных. [3,7,8]
Рекомендуется проведение ЭЭГ, ультразвукового исследования органов брюшной полости 1 раз в 3 мес, регулярное определение уровня печеночных ферментов. [3,7,8]
Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам, направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты. Перспективы развития зависят от степени пораженности хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае становится мутация), другие никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы). С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки. Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, немногие — и ночью. Некоторые люди с синдромом Ангельмана способны есть при помощи ножа и вилки, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний». Во взрослом возрасте может появиться ожирение и ухудшиться состояние позвоночника (сколиоз). Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. [3,7,8]
Девочка 3 года 1 мес. Жалобы на задержку психо- речевого, моторного развития, самостоятельно не стоит, не ходит. Беспокойный сон, приступы судорог во время сна, стереотипии, навязчивый смех, скрипит зубами во сне (бруксизм), сниженный аппетит, часто «высовывает язык». Наследственность, со слов, не отягощена. Общее состояние- удовлетворительное. Диспластический фенотип: высокий лоб с выступающими лобными буграми, уплощенная спинка носа, энофтальм, макроглоссия, тонкая верхняя губа, широкий первый палец стоп. Зубы мелкие, с диастемами. Кожные покровы обычной окраски, чистые, сыпей нет. Не лихорадит. Катаральных явлений нет. Зев спокойный. Периферические лимфоузлы не увеличены. В легких дыхание везикулярное, хрипов нет. Тоны сердца звучные, ритмичные. Живот мягкий, безболезненный при пальпации. Печень и селезенка не увеличены. Стул со склонностью к запорам. Диурез в норме. Неврологический статус: сознание ясное. Окружность головы- 48 см. (меньше 3 перцентиля). Усилен венозный рисунок в височных областях. Глазные щели симметричные, зрачки равные, фотореакции живые, симметричные, движения глазных яблок не ограничены в стороны. Косоглазие непостоянное расходящееся альтернирующее. Легкая лицевая асимметрия. Язык по средней линии, глотание не нарушено. Рот приоткрыт, гипомимия, гиперсаливация. Слух в норме. Мышечный тонус D=S, гипотония. Сухожильные рефлексы с верхних конечностей D=S, живые, с нижних конечностей D=S, живые. Опора симметричная, плоско-вальгусные стопы. Голову удерживает, сидит самостоятельно.
Самостоятельно не стоит, не ходит. Менингеальных симптомов нет. Патологические стопные знаки: отрицательные. Дермографизм красный, быстро исчезающий.
Эмоционально лабильна, улыбается, узнает близких, инструкции не выполняет, обращенную речь не понимает. Игрушками интересуется мало. Мелкая моторика рук, манипуляции с предметами снижены. Стереотипии. В активном словаре слов нет, речь на уровне голосовых реакций. Выраженная ЗПМРР. Также были зарегистрированы эпизоды ночных пароксизмов.
Анамнез: Девочка от 3-й беременности (1- 2006г.- здоровая девочка, 2- 2007г.- аборт по желанию матери), протекавшей на фоне ОРВИ, H. labialis(в 30 нед.), отеков, многоводия, самостоятельных родов на 40 неделе с массой тела- 3700 гр., длиной- 53 см., оценкой по шкале Апгар 9/9 баллов. Длительная конъюгационная желтуха (прошла в 1мес.). С рождения мама отмечала, что девочка долго ест и много срыгивает, большой язык (часто высовывала его изо рта). С рождения отмечалась гипотония. До 1 года развивалась с задержкой- голову начала держать в 7 месяцев, самостоятельно села в 1 год и 4 месяца. Наблюдается с диагнозом ДЦП. С 5 месяцев мама заметила подергивания рук 6-10 раз в сутки. Девочка смеется без видимых причин.
Отмечается склонность к запорам. Плохо ест. Периодически скрипит зубами (бруксизм), совершает жевательные движения челюстями во время беспокойства. Спит беспокойно и мало. C 04.09.2015 по 18.09.2015 находилась в неврологическом отделении ГБУЗ СК «ДКБ» г. Пятигорска, с диагнозом: ПЭП. Синдром двигательных нарушений. Гипертензионно- гидроцефалический синдром. ЭЭГ- нормального типа организации, эпилептоидной активности не выявлено, косвенные признаки гидроцефалии. НСГ- эхопризнаки дилатации желудочковой системы головного мозга. Офтальмолог- здорова. Получала Глицин, Элькар, ФТЛ. Выписана в стабильном компенсированном состоянии.
Через некоторое время после проведенного амбулаторного лечения и выполнения рекомендаций, мама вновь отметила появление подергивания рук до 7 раз в сутки при пробуждении и при засыпании, продолжительностью до 5 минут. В связи с этим находилась в неврологическом отделении ГБУЗ СК «ДКБ» г. Пятигорска, с диагнозом: Симптоматическая эпилептическая энцефалопатия. Атонически- астатический синдром с грубым отставанием двигательного и психического развития (с- м ДЦП). Гидроцефальный синдром в стадии компенсации. ЭЭГ- выраженная генерализованная гиперсинхронная тета-активность. НСГ- эхопризнаки дилатации желудочковой системы головного мозга. Получала Депакин- сироп, Дексаметазон в/м, фенибут. МРТ головного мозга- последствия перинатального постишемического поражения ЦНС (возможно, в том числе последствия нейроинфекции) в виде участков перивентрикулярной лейкодистрофии, лейкопатии и мелкоочаговых изменений вещества мозга резидуального и дистрофического характера; участки незавершенной миелинизации; участки изменения архитектоники кортикальных борозд и извилин полушарий мозга, умеренная асимметрия гиппокампов, на фоне нельзя полностью исключить кортикальные аномалии и дисплазии; умеренная наружная гидроцефалия заместительного характера; умеренная внутренняя гидроцефалия; умеренное кистовидное расширение субарахноидального пространства в полюсных отделах височных областей, умеренно кистовидно расширены большая цистерна мозга и мосто-мозжечковые цистерны по типу небольших арахноидальных кист. ТМС- норма.
Постоянно в наблюдении у невролога, генетика. Было проведено ТСХ углеводов в крови- выявлена глюкоза (норма); ТСХ аминокислот крови- патологии не выявлено; Кариотип- 46, ХХ (женский кариотип, норма); ТСХ аминокислот в моче- патологии не выявлено; Качественные тесты с мочой (уринализис): проба на гипераминоацидурию- отриц., тест с магниевым реактивом- выявлены данные за ацидоз, тест на цистин, гомоцистин- отриц., тест на гомогентизиновую кислоту- отриц., тест на щавелевую кислоту- отриц., проба с 2.4- ДНФГ на кетокислоты- отриц., тест Обермейера- на индикан- отриц., тест на ксантуреновую кислоту- отриц., тест на метилмалиновую кислоту- отриц., проба Бенедикта- отриц., проба Селиванова на фруктозу- отриц., проба на галактозу и лактозу- отриц., проба Рубнера- отриц., тест на пентозы с реактивом Биаля- отриц., Тандемная масс-спектрометрия- активность измеренных лизосомных ферментов (галактоцереброзидаза, а- глюкозидаза, а- галактозидаза, б- глюкоцереброзидаза, сфингомиелиназа, а- идуронидаза) в пределах референсных значений. Выписана в стабильном компенсированном состоянии. Рекомендовано обследование и лечение в ГБУЗ СК «КДКБ» г. Ставрополя.
С 24.11.2016 по 09.12.2016 находилась в психоневрологическом отделении ГБУЗ СК «КДКБ» г. Ставрополя с диагнозом: ПРОП ЦНС смешанного генеза (гипоксически- ишемического, инфекционного(?) поражения, МР- признаки лейкодистрофии, лейкопатии): выраженная задержка психо- речевого и моторного развития, ДЦП смешанная форма. Начальная резидуальная форма. Симптоматическая эпилепсия. Внутренняя гидроцефалия (МР- признаки). Диспластический фенотип. В динамике исключить наследственную патологию. Сопутствующий диагноз: гиперметропия средней степени обоих глаз. Плоско- вальгусные стопы. Дисфункция сфинктера Одди по билиарному типу, вследствие нарушений нейрогуморальной регуляции. Дефицит веса 1 ст. Острый ринофарингит. Острый бронхит. ДН 0. Исключить эндокринную патологию. Получала в/м ККБ, мексидол, per os: депакин- сироп, элькар 30%, тералиджин, per rectum: виферон 500 тыс. 2р/день, per os: сумамед, эреспал, ингаляции с физ. раствором, массаж, ЛФК, вертикализация, гашение тонических рефлектов. ЭЭГ (24.11.2016) – выраженные общемозговые изменения БЭА головного мозга. Признаки дисфункции диэнцефальных структур. УЗИ органов брюшной полости: печень в размерах не увеличена, с диффузными изменениями паренхимы. Деформация желчного пузыря. ПЖЖ, селезенка не изменена. Окулист- гиперметропия средней степени обоих глаз. Психолог- на первый план выступают признаки органического поражения ЦНС, грубая задержка психо- речевого и моторного развития. Эпилептолог- с-м Ретта? Подозрение на митохондриальное заболевание.
Ортопед- плоско- вальгусные стопы. Логопед- выраженная ЗРР. Гастроэнтеролог- Дисфункция сфинктера Одди по биллиарному типу, вследствие нарушений нейрогуморальной регуляции. Дефицит веса 1ст. Эндокринолог- исключить патологию щитовидной железы. Генетик- ПРОП ЦНС: выраженная задержка психо- речевого и моторного развития, ДЦП смешанная форма. Начальная резидуальная форма. Симптоматическая эпилептическая эпилепсия. Внутренняя гидроцефалия (КТ- признаки). Диспластический фенотип. Рекомендована консультация генетика, невролога, эпилептолога, ортопеда, окулиста, гастроэнтеролога и эндокринолога. Консультация генетика (24.05.2017): ПРОП ЦНС. Выраженная задержка психоречевого и моторного развития. ДЦП, смешанная форма. Симптоматическая эпилептическая энцефалопатия.
Внутренняя гидроцефалия по КТ. Диспластический фенотип.
Консультация детского эндокринолога (14.06.2017): Суклинический гипотиреоз. Недостаток витамина D. ЭЭГ от 02.08.2017: Значительные общемозговые изменения по органическому типу, значительная дисфункция стволовых и подкорковых структур, выраженные признаки задержки формирования корковой ритмики.
На фоне медленноволновой активности регистрируются вспышки медленных волн- в ритме тета, комплексы ОМВ с акцентом в теменно- височных и центральных областях бифронтально. Мониторинг ЭЭГ (13.08.2017): Заключение: На ЭЭГ бодрствования- основной ритм сформирован по возрасту, существенно дезорганизован медленно- волновой активностью, выраженные общемозговые изменения по органическому типу. Регистрируется периодическое дельта-замедление 2,5 Гц, амплитудой до 350 мкВ, длительностью до 4-х секунд в лобных областях, в небольшом количестве регистрируются медленные комплексы ОМВ (тета-дельта) амплитудой до 600 мкВ в центрально- теменно- задневисочных областях билатерально с амплитудным акцентом слева.
На ЭЭГ сна- ЭЭГ-паттерны физиологических стадий сна сформированы, существенно нарушены по органическому типу, плохо дифференцируются. В умеренном количестве (индекс выраженности 10-15%) регистрируется эпиактивность в виде комплексов ОМВ (тета-дельта) амплитудой до 550 мкВ в центрально- теменно- задневисочных областях и в лобных областях билатерально.
В декабре 2017 года проходила консультирование в ФГБНУ «МГНЦ» г. Москвы, предположительный диагноз: синдром Ангельмана. Рекомендовано провести определение аномального метилирования критического района хромосомы 15q11.2. 09.01.2018 диагноз- синдром Ангельмана подтвердился (Заключение: выявлены молекулярно- генетические изменения, характерные для синдрома Ангельмана (отсутствие метилированного аллеля промоторной области гена SNRPN)).
С 05.03.2018 по 21.03.2018 находилась на лечении в психоневрологическом отделении ГБУЗ СК «КДКБ» г. Ставрополя с диагнозом: синдром Ангельмана ДНК- подтвержденный. Симптоматическая эпилепсия с частыми вторично- генерализованными приступами, без ремиссии, медикаментозно резистентная форма. Атонически- астатический синдром, GFMCS III-IV уровень.
Выраженная ЗПМПП. Сенсо- моторная алалия.
Сопутствующий диагноз: косоглазие непостоянное, расходящееся альтернирующее. Салурия. Плоско- вальгусная деформация стоп. Хронически стафилококковый ринит?. Получала per os: депакин-сироп 200мг 2р/сутки, топамакс 25мг- ½ капс. утром и 1 капс. вечером, кеппра (1000 мг/таб.)- 150 мг утром, 100мг вечером, магне В6, элькар, гидроксизин, свечи «Виферон-2», занятия с психологом и логопедом, ЛФК, костюм «Фаэтон». Выписана в удовлетворительном состоянии домой по наблюдение невролога по месту жительства.
Даны рекомендации (допакин- сироп 200мг 2р/сутки постоянно, топамакс 25мг 1капс. 2р/сутки постоянно, кеппра 100мг 50мг- постепенная отмена, элькар 30% р-р для питья- 7 капель 2р/сутки (утро, обед) 1 мес., мексидол 0,125 таб.- 1/3 таб. 2р/день (утро, вечер)- 1 мес., ЭЭГ через 3-4 мес., ЛФК, плавание, костюм «Фаэтон», занятие с логопедом-дефектологом и психологом.
Таким образом, данный клинический случай наглядно иллюстрирует типичные электро-клинические особенности синдрома Ангельмана в сочетание с эпилепсией, а также демонстрируется сложность ранней диагностики микроделеционных синдромов (МДС), что связано с замедленным проявлением специфических признаков, характерных для данного синдрома.
Прогноз. Перспективы развития зависят от степени пораженности хромосомы. Тяжесть симптомов, возникающих при синдроме Ангельмана, отличаются в каждом конкретном случае.
Считается, что определенный генетический механизм, лежащий в основе расстройства, коррелирует с общим прогнозом для потерпевшего.
Так, мутации в гене UBE3A, связанны с наименьшим влиянием болезни на организм, а крупные делеции 15 хромосомы - хуже влияют на больных синдромом Ангельмана. Клинические признаки синдрома Ангельмана с возрастом меняются. Общее состояние здоровья достаточно хорошее, продолжительность жизни - средняя.
Список литературы:
1. http://rd17.ru/microsindroms
2. Иванова О.К. ХАРАКТЕРИСТИКА СИНДРОМА АНГЕЛЬМАНА // Материалы VIII Международной студенческой электронной научной конференции «Студенческий научный форум»
3. Атлас редких болезней / Науч. центр здоровья детей [и др.]; под ред. А.А. Баранова, А92 Л.С. Намазовой-Барановой. 2-е изд., испр. и доп. — М. ПедиатрЪ, 2016. — 420 с. ISBN 978-5-906332-28-8
4. Facts about Angelman syndrome. September 29, 2012.
5. Yamasaki K., et al. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet, 2003.
6. Verdecia M.A., et al..Conformational flexibility underlies ubiquilin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell, 2003.
7. Fridman C., et al., Paternal UPD15: further genetic and clinical studies in four Angelman syndrome patients. Am J Med Genet, 2000.
8. Nazlican H., et al. Somatic mosaicism in patients with Angelman syndrome and an imprinting defect. Hum Mol Genet, 2004.
9. Lossie A.C., et al..Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet, 2001.
